Rối loạn phổ viêm tủy thị thần kinh (NMOSD) là một bệnh viêm, qua trung gian kháng thể và tự miễn, ảnh hưởng đến hệ thần kinh trung ương. Trước đây được gọi là “bệnh Devic” và trong nhiều năm được coi là một phân nhóm của bệnh đa xơ cứng (MS). Tuy nhiên, sau khi phát hiện ra tự kháng thể kháng aquaporin-4 (AQP4-IgG) vào năm 2004, nó đã được xác lập như một thực thể bệnh độc lập.
Bối cảnh lịch sử: Năm 1870, Ngài Thomas Clifford Allbutt lần đầu tiên mô tả mối liên quan giữa viêm tủy sống và rối loạn thần kinh thị giác. Nghiên cứu sau đó cho thấy đây là một bệnh khác với MS, và hiện nay được hiểu theo khái niệm toàn diện “NMOSD”.
Dịch tễ học như sau:
Tỷ lệ mắc: Tỷ lệ mắc hàng năm ước tính của AQP4+NMOSD là 0,4-7,3 trên một triệu người1)
Khác biệt giới tính: Tỷ lệ giới tính khoảng 1:9, với ưu thế rõ rệt ở nữ.
Tuổi khởi phát: Chủ yếu xảy ra ở tuổi trung niên (40–60 tuổi), cao điểm ở cuối 30 đến đầu 40 tuổi.
Chủng tộc: Phổ biến hơn ở người gốc Phi và châu Á1)
Liên quan đến thai kỳ: Khoảng 20–47% phụ nữ có cơn đầu tiên trong thời kỳ mang thai hoặc trong vòng một năm sau sinh hoặc sảy thai.
QNMOSD khác với bệnh đa xơ cứng (MS) như thế nào?
A
NMOSD là bệnh qua trung gian kháng thể nhắm vào kênh nước AQP4 trên tế bào hình sao, và khác biệt cơ bản với MS về sinh lý bệnh, điều trị và tiên lượng. Trong NMOSD, LETM và viêm dây thần kinh thị giác nặng là điển hình, và một điểm khác biệt quan trọng là các thuốc điều chỉnh bệnh hiệu quả trong MS như interferon beta có thể kích hoạt tái phát ở NMOSD.
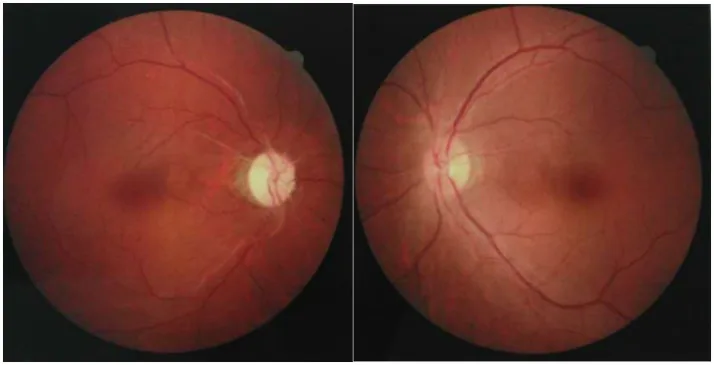
Chuai Y, et al. Paraneoplastic neuromyelitis optica spectrum disorder with dual AQP4-IgG and CRMP5 antibodies following thymectomy: a case report. Front Neurosci. 2026. Figure 1. PMCID: PMC13006573. License: CC BY.
Ảnh đáy mắt của cả hai mắt, cho thấy teo thị giác với đĩa thị nhợt nhạt ở một mắt và sưng nhẹ đĩa thị ở mắt kia. Những hình ảnh này mô tả các dấu hiệu thần kinh thị giác từ giai đoạn cấp đến mạn tính của viêm dây thần kinh thị giác trong NMOSD.
Khiếm khuyết thị trường: Không chỉ giới hạn ở ám điểm trung tâm, mà có thể bao gồm bán manh ngang, bán manh thái dương hai bên và bán manh đồng danh. Điều này là do tổn thương lan rộng đến giao thoa thị giác và dải thị giác.
Rối loạn cảm giác và liệt hai chân: Rối loạn vận động và cảm giác do viêm tủy.
Rối loạn bàng quang và trực tràng: Rối loạn thần kinh tự chủ kèm theo viêm tủy
Nấc cụt khó chịu, buồn nôn và nôn: Triệu chứng đặc trưng do tổn thương vùng postrema
Rối loạn vận động mắt: Do tổn thương thân não
Chứng ngủ nhiều (giống ngủ rũ): Do tổn thương gian não/vùng dưới đồi
Giai đoạn khởi phát có thể có triệu chứng giống cúm (sốt, đau cơ, đau đầu).
Hai bên đồng thời: Viêm dây thần kinh thị giác trong NMOSD là hai bên đồng thời ở 17-82% trường hợp. Khác biệt quan trọng với MS.
Giảm thị lực nghiêm trọng: Trong AQP4+NMOSD, trung vị thị lực thấp nhất là mức thị lực đếm ngón tay (HM). Sau hồi phục, trung vị vẫn là mức đếm ngón tay, và 60-69% có suy giảm thị lực vĩnh viễn ít nhất 20/200 ở một mắt.
Viêm tủy
LETM (Viêm tủy cắt ngang dài lan rộng): Tổn thương liên tục kéo dài ≥3 đốt sống. Khoảng 85% AQP4+NMOSD có biểu hiện này trong viêm tủy cấp.
Hội chứng tủy hoàn toàn: Liên quan đến cả ba đường vận động, cảm giác và tự chủ.
Suy giảm chức năng nặng: Hơn 30% bệnh nhân phụ thuộc xe lăn ở điểm thấp nhất của cơn. 37-44% AQP4+NMOSD cuối cùng cần dụng cụ hỗ trợ đi lại.
Hội chứng vùng postrema
Nấc cụt khó trị: Kéo dài vài ngày đến vài tuần, không đáp ứng với thuốc chống nôn thông thường.
Buồn nôn và nôn: Do vùng postrema thiếu hàng rào máu não, đây là vị trí dễ dàng tiếp cận trực tiếp của AQP4-IgG.
Dấu hiệu chẩn đoán cốt lõi của NMOSD: Nấc cụt khó trị không giải thích được là yếu tố thúc đẩy nghi ngờ tích cực NMOSD.
QViêm thần kinh thị giác trong NMOSD khác với viêm thần kinh thị giác liên quan đến MS như thế nào?
A
Viêm thần kinh thị giác trong NMOSD nặng hơn, hai bên và tái phát, với tiên lượng thị lực kém. Trong NMOSD dương tính với AQP4, 60-69% được ước tính có suy giảm thị lực vĩnh viễn ít nhất 20/200 ở một mắt. Ngoài ra, nó có xu hướng liên quan đến giao thoa thị giác, gây ra các khiếm khuyết thị trường đa dạng như bán manh thái dương hai bên, khác biệt so với MS.
MRI tủy sống: LETM là đặc trưng nhất. Ưu thế chất xám trung tâm. Kèm phù tủy, tín hiệu thấp trên T1, và ngấm thuốc Gd. Khoảng 85% bệnh nhân NMOSD AQP4+ có LETM trong viêm tủy cấp 1)
MRI thần kinh thị giác: Chuỗi xung ức chế mỡ là bắt buộc. Viêm hai bên và dài (>50%) là đặc trưng. Liên quan đến phần sau và giao thoa thị giác là điển hình của NMOSD AQP4+ 1)
MRI não: Tổn thương vùng postrema, tổn thương thân não quanh não thất IV, tổn thương vùng dưới đồi/quanh não thất III, và tổn thương chất trắng lan rộng.
Đặc điểm của NMOSD: Khác với MS, tổn thương T2 mới không triệu chứng hiếm gặp (3-13%). MRI giám sát thường không cần thiết 1)
Ở người cao tuổi, cần phân biệt với bệnh thần kinh thị giác do thiếu máu cục bộ, bệnh tủy cổ, nhồi máu tủy sống và u lympho hệ thần kinh trung ương nguyên phát.
QNMOSD có thể được chẩn đoán ngay cả khi kháng thể AQP4 âm tính không?
A
Có. Ngay cả khi AQP4-IgG âm tính hoặc chưa xét nghiệm, nếu đáp ứng hai hoặc nhiều đặc điểm lâm sàng chính, đáp ứng các yêu cầu MRI bổ sung và loại trừ các bệnh khác, NMOSD có thể được chẩn đoán. Ngoài ra, khoảng 30% trường hợp AQP4-IgG âm tính có MOG-IgG dương tính, và việc đo cả hai kháng thể được khuyến cáo. Dưới 1% trường hợp AQP4-IgG âm tính sau đó chuyển đổi huyết thanh.
Methylprednisolone 1.000 mg/ngày truyền tĩnh mạch trong 3 ngày
Nếu thị lực không cải thiện, cân nhắc lặp lại 1 đợt sau khi nghỉ 3-4 ngày
Viêm thần kinh thị giác trong NMOSD có kháng cao với steroid, do đó nếu đáp ứng không đủ, hãy cân nhắc điều trị tiếp theo sớm
Lựa chọn thứ hai: Liệu pháp trao đổi huyết tương
Được thực hiện khi không đáp ứng với steroid xung. Các lựa chọn sau đây có sẵn:
Trao đổi huyết tương đơn giản (PE): Hiệu quả cao nhất nhưng cũng gây tổn thương cơ thể nhiều nhất
Trao đổi huyết tương lọc màng kép (DFPP)
Liệu pháp hấp thụ miễn dịch (IA): Có thể loại bỏ kháng thể chọn lọc
Thứ tự hiệu quả được cho là trao đổi huyết tương đơn giản > lọc màng kép > hấp thụ miễn dịch. Thực hiện 5-6 lần mỗi đợt, và sau điều trị cần nhập viện cho đến khi lượng IgG trong cơ thể phục hồi. Lưu ý rằng đối với “viêm thần kinh thị giác” có thể không được bảo hiểm chi trả, cần giải thích cho bệnh nhân.
Trong AQP4+NMOSD, nên bắt đầu điều trị duy trì sớm sau cơn đầu tiên 1). Sau trao đổi huyết tương, thường chuyển sang prednisolone 5-10 mg/ngày + azathioprine 50-100 mg/ngày.
Các tác nhân sinh học có mức độ bằng chứng cao như sau:
Thuốc ức chế bổ thể
Eculizumab: 900 mg IV mỗi tuần × 4 lần → 1.200 mg mỗi 2 tuần như liều duy trì 1)
Ravulizumab: Liều nạp dựa trên cân nặng (2.400–3.000 mg) → sau ngày thứ 15, 3.000–3.600 mg mỗi 8 tuần1)
Liệu pháp loại bỏ tế bào B
Rituximab: 375 mg/m² IV hàng tuần × 4 lần, hoặc 1.000 mg × 2 (cách nhau 2 tuần) → 1.000 mg × 2 mỗi 6 tháng1)
Inebilizumab: 300 mg IV mỗi 15 ngày × 2 lần → mỗi 6 tháng1)
Thuốc ức chế thụ thể IL-6
Satralizumab: 120 mg tiêm dưới da, mỗi 4 tuần1)
QNếu liệu pháp steroid liều cao không hiệu quả thì sao?
A
Trao đổi huyết tương là lựa chọn tiếp theo. Có ba loại: trao đổi huyết tương đơn giản, trao đổi huyết tương qua màng lọc kép và hấp phụ miễn dịch. Trao đổi huyết tương đơn giản được coi là hiệu quả nhất nhưng cũng gây căng thẳng nhất cho cơ thể. Một đợt điều trị gồm 5–6 lần, và sau điều trị cần nhập viện.
NMOSD về cơ bản là bệnh lý tế bào hình sao (astrocytopathy). Cơ chế bệnh như sau:
Sản xuất kháng thể và vượt qua hàng rào máu não (BBB)
Ở ngoại vi, tế bào B biệt hóa thành nguyên bào plasma tiết AQP4-IgG. IL-6 thúc đẩy sự biệt hóa này và làm tăng tính thấm của hàng rào máu não. Vùng postrema là vùng thiếu hàng rào máu não và có thể là đường xâm nhập của AQP4-IgG vào hệ thần kinh trung ương.
Chuỗi tổn thương tế bào hình sao
AQP4-IgG gắn kết với các kênh nước AQP4 biểu hiện dày đặc trên chân tế bào hình sao, gây tổn thương tế bào hình sao qua các con đường sau.
Hoạt hóa con đường cổ điển của bổ thể: Phần Fc của AQP4-IgG kích hoạt bổ thể, hình thành phức hợp tấn công màng (MAC) gây tổn thương trực tiếp tế bào hình sao
ADCC (độc tế bào phụ thuộc kháng thể): Tế bào NK và bạch cầu trung tính gây tổn thương tế bào hình sao qua thụ thể Fcγ
Giải phóng C5a anaphylatoxin: Tuyển mộ bạch cầu hạt (bạch cầu trung tính, bạch cầu ái toan) gây tổn thương sợi trục thứ phát và mất myelin1)
Trong MS, mất myelin chủ yếu ở chất trắng do tế bào T CD8+ là chính, trong khi ở NMOSD, sự tham gia của tế bào T CD4+ lớn hơn, hình thành các tổn thương hoại tử ảnh hưởng đến cả chất xám và chất trắng.
Lý do phân bố
Các kênh AQP4 phân bố nhiều ở dây thần kinh thị giác, vùng postrema và tủy sống, khiến các vùng này trở thành mục tiêu chọn lọc.
Dấu ấn sinh học
GFAP huyết thanh: Phản ánh tổn thương tế bào hình sao và tăng cao trong các cơn
Neurofilament chuỗi nhẹ huyết thanh (NfL): Phản ánh tổn thương sợi trục và tương quan với mức độ nghiêm trọng của cơn1)
Các cytokine liên quan bao gồm IL-6, IL-10, IL-17a, G-CSF, TNF-α và BAFF/APRIL đã được báo cáo.
7. Nghiên cứu mới nhất và triển vọng tương lai (Báo cáo giai đoạn nghiên cứu)
Ước tính 3-5% NMOSD là cận u. Các trường hợp liên quan đến u quái buồng trứng đã được nghiên cứu chi tiết.
Ikeguchi và cộng sự (2021) đã thực hiện đánh giá 6 trường hợp NMOSD liên quan đến u quái buồng trứng có AQP4 dương tính2). Tất cả đều là nữ, tuổi khởi phát trung bình 32,7 tuổi (15-50 tuổi). Trong 6 trường hợp, 83% (5/6) có buồn nôn và nôn, 83% có dải oligoclonal dịch não tủy dương tính, và 83% có tổn thương thân não lưng. Phân tích bệnh lý xác nhận phản ứng miễn dịch AQP4 và thâm nhiễm tế bào lympho trong mô thần kinh dương tính GFAP trong khối u, gợi ý cơ chế trình diện kháng nguyên AQP4 trong khối u kích hoạt phản ứng tự miễn. Sau khi cắt bỏ khối u, AQP4-IgG chuyển sang âm tính ở 60% (3/5) trường hợp.
Ding và cộng sự (2021) đã thực hiện đánh giá 43 trường hợp NMOSD cận u3). 88,4% là nữ, với ung thư vú và ung thư phổi là các loại khối u phổ biến nhất. Tầm quan trọng của việc tầm soát khối u, đặc biệt ở bệnh nhân NMOSD trên 50 tuổi, được nhấn mạnh.
Khuyến cáo tầm soát khối u bao gồm u quái ngay cả ở bệnh nhân trẻ tuổi.
Liệu pháp hấp thụ miễn dịch cho các trường hợp kháng trị
Hiệu quả của liệu pháp hấp thụ miễn dịch Protein-A (IA) đã được báo cáo trong NMOSD kháng trị không đáp ứng với steroid, trao đổi huyết tương hoặc rituximab.
Fan và cộng sự (2024) đã thực hiện 3 buổi hấp thụ miễn dịch Protein-A trên một phụ nữ 35 tuổi mắc NMOSD liên quan đến hội chứng Sjögren kháng trị không đáp ứng với liệu pháp xung steroid và IVIG4). Trong vòng một tuần, các rối loạn thị giác, liệt hai chi dưới và rối loạn cảm giác bản thể cải thiện đáng kể, và quan sát thấy sự giảm nhanh AQP4-IgG, IgA, IgG và IgM. Không ghi nhận tái phát hoặc tiến triển trong 4 năm theo dõi.
Đặc điểm của các trường hợp có bệnh tự miễn kèm theo
Thực trạng về các bệnh tự miễn khác nhau kèm theo NMOSD đang dần được làm rõ.
Zhu và cộng sự (2025) đã báo cáo trường hợp một bé gái 14 tuổi khởi phát NMOSD lúc 11 tuổi5). AQP4-IgG dương tính, và hội chứng Sjögren nguyên phát được xác nhận trong quá trình theo dõi. Duy trì thuyên giảm bằng methylprednisolone, IVIG và mycophenolate mofetil (MMF) sau đó chuyển sang tacrolimus. Bệnh tự miễn kèm theo được báo cáo ở 20-30% trường hợp NMOSD người lớn, nhưng trường hợp này cho thấy sự kết hợp này cũng có thể xảy ra ở trẻ em5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, Suzuki M, Shimoji K, Motohashi T, Yamamoto T, Shibata N, Kitagawa K. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045. doi:10.1212/NXI.0000000000001045. PMID: 34285095. PMCID: PMC8293286.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain and behavior. 2021;11(10):e2282. doi:10.1002/brb3.2282. PMID:34520629; PMCID:PMC8553315.
Fan W, Chen X, Xiao P, Wei B, Zhang Y, Huang J, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Frontiers in immunology. 2024;15:1429405. doi:10.3389/fimmu.2024.1429405. PMID:39055718; PMCID:PMC11269126.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.