Оптиконевромиелит спектра (NMOSD) — это воспалительное, антитело-опосредованное аутоиммунное заболевание, поражающее центральную нервную систему. Ранее называлось «болезнью Девика» и долгое время считалось подтипом рассеянного склероза (РС). Однако открытие в 2004 году аутоантител к аквапорину-4 (AQP4-IgG) позволило выделить его как самостоятельную нозологическую единицу.
Историческая справка: В 1870 году сэр Томас Клиффорд Оллбатт впервые описал связь между миелитом и зрительными нарушениями. Последующие исследования признали его заболеванием, отличным от РС, и теперь оно понимается в рамках общей концепции «NMOSD».
Эпидемиология: Следующая.
Заболеваемость: Предполагаемая ежегодная заболеваемость AQP4+ NMOSD составляет 0,4–7,3 на миллион человек1)
Соотношение полов: Соотношение мужчин и женщин составляет примерно 1:9 с выраженным преобладанием женщин.
Возраст начала: преимущественно у людей среднего возраста (40–60 лет). Пик приходится на конец 30-х – начало 40-х годов.
Раса: чаще встречается у лиц африканского и азиатского происхождения1)
Связь с беременностью: примерно у 20–47% женщин первый эпизод возникает во время беременности или в течение года после родов или выкидыша.
QЧем NMOSD отличается от рассеянного склероза (РС)?
A
NMOSD – это антитело-опосредованное заболевание, направленное против водного канала AQP4 астроцитов, патогенез, лечение и прогноз которого принципиально отличаются от РС. Для NMOSD типичны LETM и тяжелый оптический неврит, а такие препараты, изменяющие течение РС, как интерферон-бета, могут провоцировать обострения NMOSD, что является важным отличием.
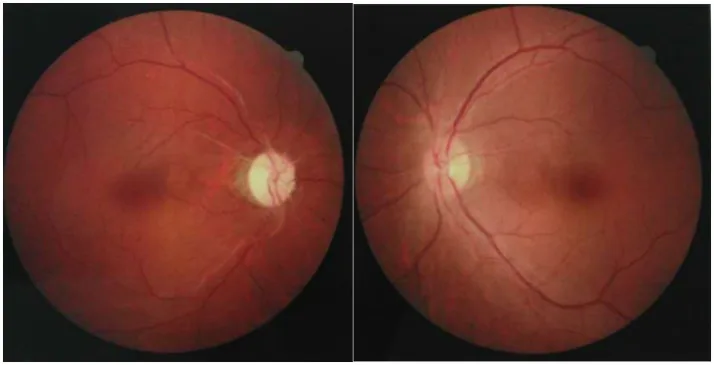
Chuai Y, et al. Paraneoplastic neuromyelitis optica spectrum disorder with dual AQP4-IgG and CRMP5 antibodies following thymectomy: a case report. Front Neurosci. 2026. Figure 1. PMCID: PMC13006573. License: CC BY.
На фотографиях глазного дна обоих глаз на одном глазу видна атрофия зрительного нерва из-за побледнения диска, на другом – легкий отек диска зрительного нерва. Представлены оптические признаки острой и хронической стадии оптического неврита при NMOSD.
Симптомы NMOSD разнообразны в зависимости от пораженной области.
Резкое снижение зрения: один из основных симптомов. Характерна устойчивость к лечению стероидами.
Боль в глазу: связана с оптическим невритом, наблюдается примерно в половине случаев.
Нарушение цветового зрения: типично снижение насыщенности красного цвета.
Дефекты поля зрения: не ограничиваются центральной скотомой, могут возникать горизонтальная гемианопсия, битемпоральная гемианопсия или гомонимная гемианопсия, так как поражение распространяется на хиазму и зрительный тракт.
Чувствительные нарушения и параплегия: двигательные и чувствительные нарушения вследствие миелита.
Нарушения функции мочевого пузыря и прямой кишки : вегетативная дисфункция, связанная с миелитом
Рефрактерная икота и тошнота/рвота : характерные симптомы, обусловленные поражением area postrema
Нарушения движений глаз : вследствие поражения ствола мозга
Гиперсомния (нарколепсиеподобная) : вследствие поражения диэнцефальной области/гипоталамуса
В начале заболевания могут наблюдаться гриппоподобные симптомы (лихорадка, миалгии, головная боль).
Отек диска зрительного нерва : наблюдается в острой фазе, впоследствии переходит в атрофию зрительного нерва.
RAPD (относительный афферентный зрачковый дефект) : одно- или двусторонний.
Двусторонний одновременный : оптический неврит при NMOSD в 17–82% случаев является двусторонним одновременным. Важное отличие от РС.
Тяжелое снижение зрения : при AQP4+ NMOSD медиана минимальной остроты зрения составляет уровень ручного счета (HM). После восстановления медиана остается на уровне счета пальцев, и у 60–69% пациентов сохраняется стойкое нарушение зрения не менее 20/200 хотя бы на одном глазу.
Миелит
LETM (продольный распространенный поперечный миелит) : непрерывное поражение протяженностью 3 и более позвонков. Примерно у 85% пациентов с AQP4+ NMOSD наблюдается при остром миелите.
Полный спинальный синдром : вовлечение всех трех путей – двигательного, чувствительного и вегетативного.
Тяжелые функциональные нарушения : более 30% пациентов прикованы к инвалидной коляске в нижней точке обострения. 37–44% пациентов с AQP4+ NMOSD в конечном итоге нуждаются в средствах для ходьбы.
Синдром area postrema
Рефрактерная икота : длится от нескольких дней до нескольких недель, не реагирует на обычные противорвотные средства.
Тошнота и рвота : из-за отсутствия гематоэнцефалического барьера в area postrema AQP4-IgG может напрямую достигать этой области.
Ключевой диагностический признак NMOSD : необъяснимая рефрактерная икота должна активно наводить на мысль о NMOSD.
QЧем отличается оптический неврит при NMOSD от такового при РС?
A
Оптический неврит при NMOSD более тяжелый, двусторонний и рецидивирующий, с худшим прогнозом для зрения. Сообщается, что у 60–69% пациентов с AQP4+ NMOSD сохраняется стойкое снижение зрения до 20/200 или хуже хотя бы на одном глазу. Кроме того, часто вовлекается хиазма, что приводит к различным дефектам поля зрения, таким как битемпоральная гемианопсия, что отличает его от РС.
МРТ спинного мозга: LETM является наиболее характерным признаком. Преимущественное поражение центрального серого вещества. Сопровождается отеком спинного мозга, гипоинтенсивностью на Т1-ВИ и накоплением Gd. Примерно 85% пациентов с AQP4+ NMOSD имеют LETM при остром миелите 1)
МРТ зрительных нервов: обязательно использование жироподавления. Характерно двустороннее, протяженное воспаление (>50%). Вовлечение задних отделов и хиазмы типично для AQP4+ NMOSD1)
МРТ головного мозга: очаги в area postrema, стволе мозга вокруг IV желудочка, гипоталамусе/перивентрикулярной области III желудочка, обширные очаги в белом веществе и др.
Особенности NMOSD: в отличие от РС, бессимптомные новые очаги на Т2-ВИ редки (3–13%). Обычно не требуется контрольная МРТ 1)
У пожилых пациентов важно дифференцировать с ишемической оптической нейропатией, шейной спондилогенной миелопатией, инфарктом спинного мозга и первичной лимфомой ЦНС
QМожет ли быть диагностирована NMOSD при отрицательных антителах к AQP4?
A
Да. Даже при отрицательных или неисследованных AQP4-IgG, если выполнены два или более основных клинических признака, дополнительные критерии МРТ и исключены другие заболевания, может быть диагностирована NMOSD. Кроме того, около 30% AQP4-IgG-отрицательных случаев являются MOG-IgG-положительными, и рекомендуется определение обоих антител. Менее 1% AQP4-IgG-отрицательных случаев впоследствии сероконвертируют.
Внутривенное введение метилпреднизолона в дозе 1000 мг/сут в течение 3 дней
При отсутствии улучшения зрения рассмотреть повторный курс после перерыва в 3–4 дня
Неврит зрительного нерва при NMOSD часто резистентен к стероидам; при недостаточном ответе следует рассмотреть следующую терапию на ранней стадии
Вторая линия: плазмаферез
Проводится при отсутствии ответа на пульс-терапию стероидами. Доступны следующие варианты:
Простой плазмаферез (ПФ) : наиболее эффективен, но также наиболее травматичен для организма
Двойной фильтрационный плазмаферез (ДФПП)
Иммуносорбция (ИА) : позволяет селективно удалять антитела
Эффективность располагается в следующем порядке: простой плазмаферез > двойная фильтрация > иммуносорбция. Один курс включает 5–6 сеансов; после лечения необходима госпитализация до восстановления уровня IgG в сыворотке. Примечание: при неврите зрительного нерва эти методы могут не покрываться страховкой, что требует разъяснения пациенту.
При AQP4+ NMOSD рекомендуется начинать поддерживающую терапию рано после первой атаки 1). После плазмафереза обычно переходят на преднизолон 5–10 мг/сут + азатиоприн 50–100 мг/сут.
Биологические препараты с высоким уровнем доказательности:
Ингибиторы комплемента
Экулизумаб : 900 мг в/в еженедельно × 4, затем 1200 мг каждые 2 недели в качестве поддерживающей дозы 1)
Равулизумаб : нагрузочная доза на основе веса (2400–3000 мг) → с 15-го дня 3000–3600 мг каждые 8 недель1)
Терапия истощения B-клеток
Ритуксимаб : 375 мг/м² в/в еженедельно × 4, или 1000 мг × 2 (с интервалом в 2 недели) → каждые 6 месяцев 1000 мг × 21)
Инебилизумаб : 300 мг в/в каждые 15 дней × 2 → каждые 6 месяцев1)
Ингибиторы рецептора IL-6
Сатрализумаб : 120 мг подкожно каждые 4 недели1)
QЧто делать, если пульс-терапия стероидами неэффективна?
A
Следующим вариантом является плазмаферез. Выбирают из трех типов: простой плазмаобмен, двойная фильтрационная плазмаферез и иммуноадсорбция. Простой плазмаобмен считается наиболее эффективным, но также наиболее нагрузочным для организма. Проводится курс из 5–6 сеансов, после лечения требуется госпитализация.
6. Патофизиология и детальные механизмы развития заболевания
NMOSD по своей сути является астроцитопатией. Механизм развития заболевания следующий:
Продукция антител и прохождение через гематоэнцефалический барьер (ГЭБ)
На периферии B-клетки дифференцируются в плазмобласты, секретирующие AQP4-IgG. IL-6 способствует этой дифференцировке и повышает проницаемость гематоэнцефалического барьера (ГЭБ). Area postrema — область, лишенная ГЭБ, и может служить путем проникновения AQP4-IgG в ЦНС.
Каскад повреждения астроцитов
AQP4-IgG связываются с водными каналами AQP4, высоко экспрессированными на ножках астроцитов, что приводит к повреждению астроцитов по следующим путям.
Активация классического пути комплемента: Fc-часть AQP4-IgG активирует комплемент, образуя мембраноатакующий комплекс (MAC), который напрямую повреждает астроциты.
ADCC (антителозависимая клеточная цитотоксичность): NK-клетки и нейтрофилы повреждают астроциты через Fcγ-рецепторы.
При РС доминирует CD8+ T-клеточно-опосредованная демиелинизация белого вещества, тогда как при NMOSD большее участие принимают CD4+ T-клетки, формируя некротические поражения как серого, так и белого вещества.
Причина распределения
Каналы AQP4 обильно распределены в зрительном нерве, area postrema и спинном мозге, поэтому эти области избирательно поражаются.
Биомаркеры
Сывороточный GFAP: Отражает повреждение астроцитов и повышен во время приступа.
Сывороточная легкая цепь нейрофиламентов (NfL): Отражает повреждение аксонов и коррелирует с тяжестью приступа1)
Вовлеченные цитокины включают IL-6, IL-10, IL-17a, G-CSF, TNF-α и BAFF/APRIL.
7. Новейшие исследования и перспективы на будущее (отчёты на стадии исследований)
По оценкам, 3–5% случаев NMOSD являются паранеопластическими. Особенно подробно изучены случаи, связанные с тератомой яичника.
Ikeguchi и соавт. (2021) провели обзор 6 случаев AQP4+ NMOSD, ассоциированного с тератомой яичника 2). Все пациенты — женщины, средний возраст начала 32,7 года (15–50 лет). Из 6 случаев у 83% (5/6) наблюдались тошнота/рвота, у 83% — положительные олигоклональные полосы в ЦСЖ, у 83% — поражения дорсального отдела ствола мозга. Патологический анализ подтвердил AQP4-иммунореактивность и лимфоцитарную инфильтрацию в GFAP-положительной нервной ткани внутри опухоли, что предполагает механизм, при котором презентация антигена AQP4 в опухоли запускает аутоиммунную реакцию. После удаления опухоли AQP4-IgG стал отрицательным у 60% (3/5) пациентов.
Ding и соавт. (2021) провели обзор 43 случаев паранеопластического NMOSD3). 88,4% были женщинами, наиболее частыми типами опухолей были рак молочной железы и рак лёгких. Особо подчёркивается важность скрининга опухолей у пациентов с NMOSD в возрасте 50 лет и старше.
Скрининг на опухоли, включая тератомы, рекомендуется даже у молодых пациентов.
Сообщается об эффективности иммуноадсорбции на протеине A (ИА) для рефрактерного NMOSD, не отвечающего на стероиды, плазмаферез или ритуксимаб.
Fan и соавт. (2024) провели 3 сеанса иммуноадсорбции на протеине A у 35-летней женщины с рефрактерным NMOSD в сочетании с синдромом Шёгрена, не отвечавшим на пульс-терапию стероидами и ВВИГ 4). В течение недели значительно улучшились нарушения зрения, параплегия и проприоцептивные нарушения, и было отмечено быстрое снижение AQP4-IgG, IgA, IgG и IgM. За 4 года наблюдения рецидивов или прогрессирования не наблюдалось.
Особенности случаев с сочетанием аутоиммунных заболеваний
Реальность сочетания различных аутоиммунных заболеваний с NMOSD становится всё более ясной.
Zhu и соавт. (2025) сообщили о случае 14-летней девочки, у которой развился NMOSD в возрасте 11 лет 5). AQP4-IgG-положительный, в ходе течения было подтверждено сочетание с первичным синдромом Шёгрена. Ремиссия поддерживалась метилпреднизолоном, ВВИГ и микофенолата мофетилом (ММФ) с последующим переходом на такролимус. Хотя у взрослых пациентов с NMOSD сообщается о сочетании с аутоиммунными заболеваниями в 20–30% случаев, было показано, что такое сочетание существует и у детей 5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, Suzuki M, Shimoji K, Motohashi T, Yamamoto T, Shibata N, Kitagawa K. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045. doi:10.1212/NXI.0000000000001045. PMID: 34285095. PMCID: PMC8293286.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain and behavior. 2021;11(10):e2282. doi:10.1002/brb3.2282. PMID:34520629; PMCID:PMC8553315.
Fan W, Chen X, Xiao P, Wei B, Zhang Y, Huang J, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Frontiers in immunology. 2024;15:1429405. doi:10.3389/fimmu.2024.1429405. PMID:39055718; PMCID:PMC11269126.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.