視神経炎

視神経脊髄炎(NMOSD)
1. 視神経脊髄炎(NMOSD)とは
Section titled “1. 視神経脊髄炎(NMOSD)とは”視神経脊髄炎スペクトラム障害(NMOSD)は、中枢神経系を侵す炎症性・抗体介在性・自己免疫疾患である。以前は「デビック病」とも呼ばれ、長年にわたり多発性硬化症(MS)の亜型と見なされてきた。しかし2004年にアクアポリン4(AQP4)に対する自己抗体(AQP4-IgG)が発見されたことで、独立した疾患単位として確立した。
歴史的背景として、1870年にSir Thomas Clifford Allbuttが脊髄炎と視神経障害の関連性を初めて記述した。その後の研究でMSとは異なる疾患と認識され、現在では「NMOSD」という包括的概念で捉えられている。
疫学は以下の通りである。
- 発生率:AQP4+NMOSDの推定年間発生率は0.4〜7.3/百万人1)
- 性差:男女比は約1:9と女性に著しく偏る
- 発症年齢:主に中年(40〜60歳)に好発。30歳代後半〜40歳代前半にピーク
- 人種:アフリカ系・アジア系に多い傾向1)
- 妊娠との関連:女性の約20〜47%が妊娠中または出産・流産から1年以内に初発する
Q
NMOSDと多発性硬化症(MS)はどう違いますか?
2. 主な症状と臨床所見
Section titled “2. 主な症状と臨床所見”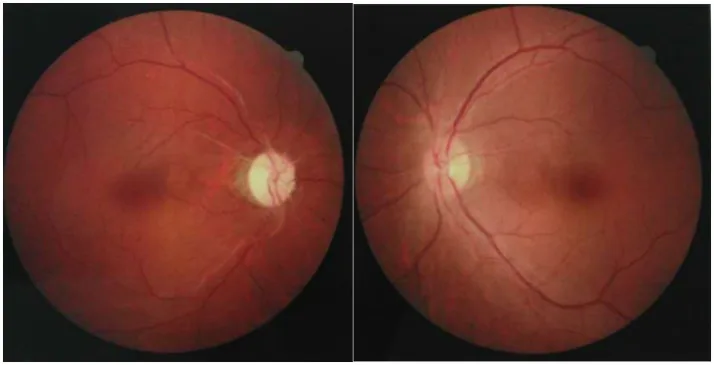
Chuai Y, et al. Paraneoplastic neuromyelitis optica spectrum disorder with dual AQP4-IgG and CRMP5 antibodies following thymectomy: a case report. Front Neurosci. 2026. Figure 1. PMCID: PMC13006573. License: CC BY.
両眼の眼底写真で、片眼に視神経乳頭の蒼白化による視神経萎縮、対側眼に軽度の視神経乳頭腫脹がみられる。NMOSDでみられる視神経炎の急性期から慢性期の視神経所見が描出される。
NMOSDの症状は侵される部位によって多様である。
- 急激な視力低下:主要症状のひとつ。ステロイド治療への抵抗性が特徴的である
- 眼痛:視神経炎に伴い、約半数の症例に認められる
- 色覚異常:赤色飽和度の低下が典型的
- 視野障害:中心暗点にとどまらず、水平半盲・両耳側半盲・同名半盲をきたすこともある。視交叉・視索にまで病変が及ぶためである
- 感覚障害・対麻痺:脊髄炎による運動・感覚障害
- 膀胱直腸障害:脊髄炎に伴う自律神経障害
- 難治性吃逆・悪心嘔吐:最後野(area postrema)の病変による特徴的症状
- 眼球運動障害:脳幹病変によるもの
- 過眠症(ナルコレプシー様):間脳・視床下部病変による
発症初期にインフルエンザ様症状(発熱・筋肉痛・頭痛)を呈することがある。
脊髄炎
LETM(長大横断性脊髄炎):3椎体以上にわたる連続病変。AQP4+NMOSDの約85%が急性脊髄炎時に示す。
完全脊髄症候群:運動・感覚・自律神経の3経路すべてを巻き込む。
重度の機能障害:30%以上の患者が発作の最低点で車椅子依存。AQP4+NMOSDの37〜44%が最終的に歩行補助具を必要とする。
最後野症候群
難治性吃逆:数日〜数週間持続し、通常の制吐薬に反応しない。
悪心・嘔吐:最後野が血液脳関門を欠くため、AQP4-IgGが直接到達しやすい部位であることが背景にある。
NMOSDの診断核心所見:説明のつかない難治性吃逆はNMOSDを積極的に疑う契機となる。
Q
NMOSDの視神経炎はMS関連のものとどう違いますか?
A
NMOSDの視神経炎はより重症・両側性・再発性であり、視力予後が不良である。AQP4+NMOSDでは60〜69%が少なくとも片眼で20/200以下の永続的視力障害を残すとされている。また視交叉を巻き込みやすく、両耳側半盲など多様な視野障害をきたす点もMSとの違いである。
3. 原因とリスク要因
Section titled “3. 原因とリスク要因”NMOSDの正確な病因は完全には解明されていない。自己免疫寛容の喪失が根本にあると考えられている。
主なリスク要因は以下の通りである。
- 女性:男女比約1:9と圧倒的に女性に多い
- 人種:アジア系・アフリカ系で発症リスクが高い
- 自己免疫疾患の合併:全身性エリテマトーデス(SLE)、シェーグレン症候群、重症筋無力症など
- NMOSDの10〜30%にシェーグレン症候群が合併する。小児例でも報告されている5)
- 重症筋無力症との合併は2〜3%に認められる1)
- 悪性腫瘍(傍腫瘍性NMOSD):NMOSDの3〜5%が傍腫瘍性と推定される2)3)
- 乳癌・肺癌・卵巣奇形腫などが報告されている3)
- 腫瘍内のAQP4発現が自己免疫反応を誘発するという機序が提唱されている2)
- 奇形腫関連NMOSDは若年女性(平均32.7歳)に多い2)
4. 診断と検査方法
Section titled “4. 診断と検査方法”診断基準(2015年国際コンセンサス)
Section titled “診断基準(2015年国際コンセンサス)”AQP4-IgG陽性NMOSDの診断基準は以下の3項目を満たすことである。
- 少なくとも1つの主要臨床的特徴
- AQP4-IgG陽性(最善の検出法使用)
- 他の診断の除外
AQP4-IgG陰性または未検査のNMOSDの診断基準は以下の4項目を満たすことである。
- 少なくとも2つの主要臨床的特徴(うち1つは視神経炎・LETM・または最後野症候群)
- 空間的多発性
- 追加MRI要件を満たす
- 他の診断の除外
**主要臨床的特徴(6項目)**は以下の通りである。
- 視神経炎
- 急性脊髄炎
- 最後野症候群(難治性吃逆・悪心嘔吐)
- 急性脳幹症候群
- 症候性ナルコレプシー/急性間脳症候群
- 症候性大脳症候群
血清学的検査
Section titled “血清学的検査”以下の表に主要な抗体検査法の比較を示す。
| 検査法 | 感度 | 特異度 | 備考 |
|---|---|---|---|
| CBA(細胞ベースアッセイ) | 69.7〜100% | 85.8〜100% | 推奨法 |
| ELISA法 | CBAよりやや劣る | CBAよりやや劣る | 日本で保険収載 |
- AQP4-IgG:NMOSDに疾患特異的。急性発作時・免疫抑制療法開始前の測定が推奨される1)
- CBA(細胞ベースアッセイ):現在の推奨検出法。ELISAの偽陽性率はCBAの5倍とされる1)
- MOG-IgG:AQP4-IgG陰性NMOSDの約30%で陽性1)
- CSFオリゴクローナルバンド(OCB):NMOSDでは10〜20%と低率(MSでは88%)。陰性はNMOSDを示唆する1)
- CSF白血球数:50/μL超・好中球・好酸球の存在はNMOSDをMSから区別する手がかりとなる
- 限界フリッカ値(CFF):視神経炎の活動性評価に有用で、NMOSDでは低下を認める
画像診断(MRI)
Section titled “画像診断(MRI)”- 脊髄MRI:LETMが最も特徴的。中央灰白質優位。脊髄腫脹・T1低信号・Gd造影効果を伴う。AQP4+NMOSDの約85%が急性脊髄炎時にLETMを呈する1)
- 視神経MRI:脂肪抑制画像が必須。両側性・長範囲の炎症(50%以上)が特徴。後方部分・視交叉の関与がAQP4+NMOSDに特徴的1)
- 脳MRI:最後野病変・第4脳室周囲脳幹病変・視床下部/第3脳室周囲病変・広範白質病変などを認める
- NMOSDの特徴:MSとは異なり、無症候性の新規T2病変は稀(3〜13%)。サーベイランスMRIは通常不要1)
- 多発性硬化症(MS)
- MOG抗体関連疾患(MOGAD)
- 急性散在性脳脊髄炎(ADEM)
- 全身性エリテマトーデス
- 神経ベーチェット病
- 高齢者では虚血性視神経症・頸椎症性脊髄症・脊髄梗塞・原発性CNSリンパ腫との鑑別が重要
Q
AQP4抗体が陰性でもNMOSDと診断されることはありますか?
A
ある。AQP4-IgG陰性または未検査の場合でも、主要臨床的特徴2つ以上を満たし、追加MRI要件を充足し、他疾患を除外できればNMOSDと診断される。また、AQP4-IgG陰性例の約30%ではMOG-IgGが陽性であり、両抗体の測定が推奨される。AQP4-IgG陰性例のうち後にセロコンバートするのは1%未満とされている。
5. 標準的な治療法
Section titled “5. 標準的な治療法”第一選択:ステロイドパルス療法
- メチルプレドニゾロン1,000mg/日の点滴静注を3日間施行する
- 視力改善がなければ中3〜4日あけて再度1クール施行を検討する
- NMOSDの視神経炎はステロイドへの抵抗性が高いため、反応不十分な場合は早期に次の治療を考慮する
第二選択:血漿交換療法
ステロイドパルスに反応しない場合に実施する。以下の方法が選択肢となる。
- 単純血漿交換療法(PE):効果は最も高いが生体へのダメージも最大
- 二重膜濾過血漿交換療法(DFPP)
- 免疫吸着療法(IA):抗体選択的除去が可能
効果の順は単純血漿交換 > 二重膜濾過 > 免疫吸着とされる。1クール5〜6回施行し、治療後は体内IgG量の回復まで入院管理が必要である。なお「視神経炎」に対しては保険適用外となる場合があり、患者への説明が必要である。
再発予防(維持療法)
Section titled “再発予防(維持療法)”AQP4+NMOSDでは初回発作後から維持治療を早期開始すべきとされる1)。血漿交換後にプレドニゾロン5〜10mg/日+アザチオプリン50〜100mg/日への移行が一般的である。
エビデンスレベルの高い生物学的製剤は以下の通りである。
補体阻害薬
- エクリズマブ:900mg IV 毎週×4回→1,200mg 2週間ごとの維持投与1)
- ラブリズマブ:体重ベースの負荷量(2,400〜3,000mg)→15日目以降3,000〜3,600mg を8週ごと1)
B細胞除去療法
- リツキシマブ:375mg/m² IV 毎週×4回、または1,000mg×2回(2週間隔)→6か月ごとに1,000mg×2回1)
- イネビリズマブ:300mg IV 15日ごとに2回→6か月ごと1)
IL-6受容体阻害薬
- サトラリズマブ:120mg 皮下注射、4週間ごと1)
Q
ステロイドパルス療法が効かない場合はどうしますか?
A
血漿交換療法が次の選択肢となる。単純血漿交換・二重膜濾過血漿交換・免疫吸着の3種類から選択する。単純血漿交換が最も効果が高いとされるが生体への負担も大きい。1クール5〜6回施行し、治療後は入院管理が必要となる。
6. 病態生理学・詳細な発症機序
Section titled “6. 病態生理学・詳細な発症機序”NMOSDは本質的にはアストロサイト病(astrocytopathy)である。発症機序は以下の通りである。
抗体産生と血液脳関門(BBB)通過
末梢でB細胞がAQP4-IgG分泌プラスマブラストに分化する。IL-6はこの分化を促進し、BBBの透過性を亢進させる。最後野はBBBを欠く領域であり、AQP4-IgGがCNSへ侵入する経路となりうる。
アストロサイト障害の連鎖
アストロサイト足突起に高密度に発現するAQP4水チャネルにAQP4-IgGが結合し、以下の経路でアストロサイト傷害が生じる。
- 補体古典経路の活性化:AQP4-IgGのFc部分が補体を活性化し、膜傷害複合体(MAC)を形成してアストロサイトを直接傷害する
- ADCC(抗体依存性細胞傷害):NK細胞や好中球がアストロサイトをFcγ受容体を介して傷害する
- C5aアナフィラトキシン放出:顆粒球(好中球・好酸球)を動員し、二次的な軸索損傷と脱髄を引き起こす1)
MSとの病理的差異
MSではCD8+ T細胞が中心的役割を担う白質中心の脱髄が主体であるが、NMOSDではCD4+ T細胞の関与が大きく、灰白質・白質の両方に及ぶ壊死性病変を形成する。
分布の理由
AQP4チャネルは視神経・最後野・脊髄に多く分布しており、これらの領域が選択的に標的となる。
バイオマーカー
- 血清GFAP:アストロサイト障害を反映し、発作時に高値となる
- 血清ニューロフィラメント軽鎖(NfL):軸索損傷を反映し、発作の重症度と相関する1)
関与するサイトカインとして IL-6・IL-10・IL-17a・G-CSF・TNF-α・BAFF/APRIL が報告されている。
7. 最新の研究と今後の展望(研究段階の報告)
Section titled “7. 最新の研究と今後の展望(研究段階の報告)”傍腫瘍性NMOSDの機序解明と腫瘍スクリーニング
Section titled “傍腫瘍性NMOSDの機序解明と腫瘍スクリーニング”NMOSDの3〜5%が傍腫瘍性と推定される。卵巣奇形腫との関連例が特に詳しく研究されている。
Ikeguchiら(2021)は卵巣奇形腫関連AQP4+NMOSDの6例レビューを実施した2)。全例女性、平均発症年齢32.7歳(15〜50歳)。6例のうち83%(5/6例)が悪心・嘔吐を呈し、83%でCSFオリゴクローナルバンド陽性、83%に背側脳幹病変を認めた。病理解析では腫瘍内のGFAP陽性神経組織にAQP4免疫反応性とリンパ球浸潤が確認され、腫瘍内でのAQP4抗原提示が自己免疫反応を惹起する機序が示唆された。腫瘍摘出後には60%(3/5例)でAQP4-IgGが陰転化した。
Dingら(2021)は43例の傍腫瘍性NMOSDのレビューを実施した3)。88.4%が女性で、乳癌と肺癌が最多の腫瘍型であった。特に50歳以上のNMOSD患者では腫瘍スクリーニングの重要性が強調されている。
若年例であっても奇形腫を含む腫瘍のスクリーニングが推奨される。
難治例に対する免疫吸着療法
Section titled “難治例に対する免疫吸着療法”ステロイド・血漿交換・リツキシマブなどに不応性の難治性NMOSDに対して、Protein-A免疫吸着療法(IA)の有効性が報告されている。
Fanら(2024)は、ステロイドパルスおよびIVIGに反応しない難治性シェーグレン症候群合併NMOSDの35歳女性に対し、Protein-A免疫吸着療法3セッションを実施した4)。1週間以内に視力障害・対麻痺・固有感覚障害が著明に改善し、AQP4-IgG・IgA・IgG・IgMの急速な低下が確認された。4年間の経過観察で再発・進行は認めていない。
自己免疫疾患との合併例の特徴
Section titled “自己免疫疾患との合併例の特徴”NMOSDへの各種自己免疫疾患合併の実態が明らかになりつつある。
Zhuら(2025)は、11歳時にNMOSDを発症した14歳女児の症例を報告した5)。AQP4-IgG陽性で、経過中に原発性シェーグレン症候群の合併が確認された。メチルプレドニゾロン・IVIG・ミコフェノール酸モフェチル(MMF)→タクロリムスへの変更により寛解を維持している。成人NMOSD症例の20〜30%に自己免疫疾患合併が報告されているが、小児例での合併も存在することが示された5)。
8. 参考文献
Section titled “8. 参考文献”- Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment: A Tale of Two Central Nervous System Autoimmune Inflammatory Disorders. Neurol Clin. 2024;42(1):77-114. doi:10.1016/j.ncl.2023.06.009. PMID:37980124; PMCID:PMC10658081.
- Ikeguchi R, Shimizu Y, Shimomura A, Suzuki M, Shimoji K, Motohashi T, Yamamoto T, Shibata N, Kitagawa K. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045. doi:10.1212/NXI.0000000000001045. PMID: 34285095. PMCID: PMC8293286.
- Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain and behavior. 2021;11(10):e2282. doi:10.1002/brb3.2282. PMID:34520629; PMCID:PMC8553315.
- Fan W, Chen X, Xiao P, Wei B, Zhang Y, Huang J, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Frontiers in immunology. 2024;15:1429405. doi:10.3389/fimmu.2024.1429405. PMID:39055718; PMCID:PMC11269126.
- Zhu GQ, Hu RX, Peng Y, Yao Y, Li GM. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Frontiers in immunology. 2025;16:1559825. doi:10.3389/fimmu.2025.1559825. PMID:40698091; PMCID:PMC12279711.