اضطراب طيف التهاب النخاع والعصب البصري (NMOSD) هو مرض التهابي، بوساطة الأجسام المضادة، ومناعي ذاتي يصيب الجهاز العصبي المركزي. كان يُعرف سابقًا باسم “داء ديفيك”، واعتُبر لسنوات طويلة نوعًا فرعيًا من التصلب المتعدد (MS). ومع ذلك، بعد اكتشاف الأجسام المضادة الذاتية لمستقبلات أكوابورين-4 (AQP4-IgG) في عام 2004، تم تأسيسه ككيان مرضي مستقل.
الخلفية التاريخية: في عام 1870، وصف السير توماس كليفورد ألبوت العلاقة بين التهاب النخاع الشوكي واضطرابات العصب البصري لأول مرة. أظهرت الأبحاث اللاحقة أنه مرض مختلف عن التصلب المتعدد، ويُفهم الآن ضمن المفهوم الشامل “NMOSD”.
الوبائيات هي كما يلي:
معدل الإصابة: يقدر معدل الإصابة السنوي لـ AQP4+NMOSD بـ 0.4-7.3 لكل مليون شخص1)
الفروق بين الجنسين: نسبة الجنس حوالي 1:9، مع غلبة واضحة للإناث.
عمر البداية: يحدث بشكل رئيسي في منتصف العمر (40-60 عامًا)، مع ذروة في أواخر الثلاثينيات إلى أوائل الأربعينيات.
العرق: أكثر شيوعًا لدى الأشخاص من أصل أفريقي وآسيوي1)
الارتباط بالحمل: حوالي 20-47% من النساء يعانين من النوبة الأولى أثناء الحمل أو خلال عام واحد من الولادة أو الإجهاض.
Qما الفرق بين التهاب النخاع والعصب البصري (NMOSD) والتصلب المتعدد (MS)؟
A
NMOSD هو مرض بوساطة الأجسام المضادة يستهدف قنوات الماء AQP4 في الخلايا النجمية، ويختلف جوهريًا عن التصلب المتعدد (MS) في الفيزيولوجيا المرضية والعلاج والتشخيص. في NMOSD، تكون الآفات الطولية الشاملة (LETM) والتهاب العصب البصري الشديد نموذجية، ومن المهم أيضًا أن الأدوية المعدلة للمرض الفعالة في MS مثل الإنترفيرون بيتا قد تحفز الانتكاسات في NMOSD.
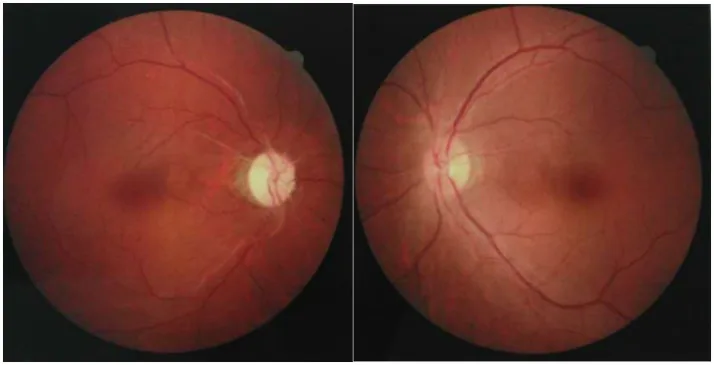
Chuai Y, et al. Paraneoplastic neuromyelitis optica spectrum disorder with dual AQP4-IgG and CRMP5 antibodies following thymectomy: a case report. Front Neurosci. 2026. Figure 1. PMCID: PMC13006573. License: CC BY.
صور قاع العين لكلتا العينين، تظهر ضمور العصب البصري مع شحوب القرص البصري في عين واحدة وتورم خفيف في القرص البصري في العين المقابلة. تُظهر هذه الصور نتائج العصب البصري في المراحل الحادة والمزمنة من التهاب العصب البصري المرتبط بـ NMOSD.
انخفاض حاد في الرؤية: أحد الأعراض الرئيسية. يتميز بمقاومة العلاج بالستيرويدات.
ألم العين: يحدث مع التهاب العصب البصري في حوالي نصف الحالات.
شذوذ رؤية الألوان: انخفاض تشبع اللون الأحمر هو النمط النموذجي.
اضطرابات المجال البصري: لا تقتصر على العتمة المركزية، بل قد تشمل عمى نصفي أفقي، عمى نصفي صدغي مزدوج، وعمى نصفي متجانس. وذلك لأن الآفة تمتد إلى التصالب البصري والسبيل البصري.
اضطرابات حسية وشلل سفلي: اضطرابات حركية وحسية ناتجة عن التهاب النخاع.
RAPD (عجز حدقي وارد نسبي): يُلاحظ في جهة واحدة أو كلتا الجهتين.
الإصابة الثنائية المتزامنة: التهاب العصب البصري في NMOSD يكون ثنائيًا متزامنًا في 17-82% من الحالات. فرق مهم عن التصلب المتعدد.
انخفاض حاد في حدة البصر: في AQP4+NMOSD، متوسط أدنى حدة بصر هو مستوى حركة اليد (HM). حتى بعد التعافي، متوسط حدة البصر هو عد الأصابع، و60-69% يبقى لديهم ضعف بصري دائم لا يقل عن 20/200 في عين واحدة على الأقل.
التهاب النخاع
LETM (التهاب النخاع المستعرض الطويل الممتد): آفة مستمرة تمتد على ثلاثة فقرات أو أكثر. حوالي 85% من حالات AQP4+NMOSD تظهر ذلك أثناء التهاب النخاع الحاد.
متلازمة النخاع الكامل: تشمل المسارات الحركية والحسية واللاإرادية الثلاثة.
إعاقة وظيفية شديدة: أكثر من 30% من المرضى يعتمدون على الكرسي المتحرك في أسوأ حالات النوبة. 37-44% من AQP4+NMOSD يحتاجون في النهاية إلى مساعدات المشي.
متلازمة المنطقة الأخيرة
الحازوقة المستعصية: تستمر لعدة أيام إلى أسابيع ولا تستجيب لمضادات القيء المعتادة.
الغثيان والقيء: نظرًا لأن المنطقة الأخيرة تفتقر إلى الحاجز الدموي الدماغي، فهي موقع يسهل وصول AQP4-IgG إليه مباشرة.
النتائج التشخيصية الأساسية لـ NMOSD: الحازوقة المستعصية غير المبررة هي مؤشر للاشتباه النشط في NMOSD.
Qكيف يختلف التهاب العصب البصري في NMOSD عن ذلك المرتبط بالتصلب المتعدد؟
A
التهاب العصب البصري في NMOSD أكثر شدة وثنائيًا ومتكررًا، ويكون تشخيص الرؤية سيئًا. في NMOSD الموجب لـ AQP4، يُقدر أن 60-69% يعانون من ضعف بصري دائم لا يقل عن 20/200 في عين واحدة على الأقل. كما أنه يميل إلى إصابة التصالب البصري، مما يسبب عيوبًا متنوعة في المجال البصري مثل العمى الشقي الصدغي، وهو اختلاف عن التصلب المتعدد.
التصوير بالرنين المغناطيسي للحبل الشوكي: التهاب النخاع المستعرض الطويل (LETM) هو الأكثر تميزًا. يغلب على المادة الرمادية المركزية. يصاحبه تورم في الحبل الشوكي، إشارة منخفضة في T1، وتعزيز بالغادولينيوم. حوالي 85% من مرضى NMOSD المصابين بـ AQP4+ يظهرون LETM أثناء التهاب النخاع الحاد 1)
التصوير بالرنين المغناطيسي للعصب البصري: تسلسل كبت الدهون ضروري. يتميز بالتهاب ثنائي الجانب وطويل المدى (أكثر من 50%). إصابة الجزء الخلفي والتصالبة البصرية مميزة لـ AQP4+ NMOSD 1)
التصوير بالرنين المغناطيسي للدماغ: آفات المنطقة الخلفية، آفات جذع الدماغ حول البطين الرابع، آفات تحت المهاد/حول البطين الثالث، وآفات المادة البيضاء الواسعة.
خصائص NMOSD: على عكس التصلب المتعدد، الآفات الجديدة غير العرضية في T2 نادرة (3-13%). عادة لا تكون هناك حاجة للتصوير بالرنين المغناطيسي للمراقبة 1)
عند كبار السن، من المهم التفريق بين الاعتلال العصبي البصري الإقفاري، اعتلال النخاع العنقي، احتشاء النخاع، ولمفوما الجهاز العصبي المركزي الأولية.
Qهل يمكن تشخيص NMOSD حتى لو كانت الأجسام المضادة لـ AQP4 سلبية؟
A
نعم. حتى في حالة سلبية AQP4-IgG أو عدم الفحص، إذا تم استيفاء اثنين أو أكثر من السمات السريرية الرئيسية، واستيفاء متطلبات التصوير بالرنين المغناطيسي الإضافية، واستبعاد الأمراض الأخرى، يمكن تشخيص NMOSD. أيضًا، حوالي 30% من الحالات السلبية لـ AQP4-IgG تكون إيجابية لـ MOG-IgG، ويوصى بقياس كلا الجسمين المضادين. أقل من 1% من الحالات السلبية لـ AQP4-IgG تتحول لاحقًا إلى إيجابية.
إعطاء ميثيل بريدنيزولون 1000 ملغ/يوم عن طريق الوريد لمدة 3 أيام
إذا لم يتحسن البصر، يُنظر في تكرار دورة واحدة بعد فترة 3-4 أيام
بما أن التهاب العصب البصري في NMOSD مقاوم للستيرويدات، يجب النظر في العلاج التالي مبكرًا إذا كانت الاستجابة غير كافية
الخيار الثاني: علاج تبادل البلازما
يتم إجراؤه في حالة عدم الاستجابة للستيرويد النبضي. الخيارات التالية متاحة:
تبادل البلازما البسيط (PE): الأكثر فعالية ولكنه الأكثر ضررًا للجسم
تبادل البلازما بالترشيح المزدوج (DFPP)
العلاج بالامتصاص المناعي (IA): إزالة انتقائية للأجسام المضادة
يُعتقد أن ترتيب الفعالية هو تبادل البلازما البسيط > الترشيح المزدوج > الامتصاص المناعي. يتم إجراء 5-6 جلسات كدورة واحدة، وبعد العلاج يلزم الإقامة في المستشفى حتى يتعافى مستوى IgG في الجسم. تجدر الإشارة إلى أنه قد لا يكون مشمولاً بالتأمين لعلاج “التهاب العصب البصري”، مما يتطلب شرحًا للمريض.
في AQP4+NMOSD، يُوصى ببدء العلاج المداوم مبكرًا بعد النوبة الأولى 1). بعد تبادل البلازما، من الشائع التحول إلى بريدنيزولون 5-10 ملغ/يوم + أزاثيوبرين 50-100 ملغ/يوم.
العوامل البيولوجية ذات مستوى الأدلة العالية هي كما يلي:
مثبطات المتممة
إيكوليزوماب: 900 ملغ وريدياً أسبوعياً × 4 مرات → 1200 ملغ كل أسبوعين كجرعة مداومة 1)
رافليزوماب: جرعة تحميل تعتمد على الوزن (2400-3000 مجم) → بعد اليوم 15، 3000-3600 مجم كل 8 أسابيع1)
إينيبيليزوماب: 300 مجم وريدياً كل 15 يوماً مرتين → كل 6 أشهر1)
مثبطات مستقبل IL-6
ساتراليزوماب: 120 مجم تحت الجلد، كل 4 أسابيع1)
Qماذا لو لم يستجب المريض للعلاج بالستيرويد بجرعات عالية؟
A
تبادل البلازما هو الخيار التالي. يتم الاختيار من بين ثلاثة أنواع: تبادل البلازما البسيط، وتبادل البلازما بغشاء مزدوج، والامتصاص المناعي. يُعتبر تبادل البلازما البسيط الأكثر فعالية ولكنه أيضاً الأكثر إجهاداً للجسم. يتم إجراء 5-6 جلسات كدورة واحدة، ويتطلب العلاج دخول المستشفى.
NMOSD هو في الأساس مرض الخلايا النجمية (astrocytopathy). آلية المرض هي كما يلي:
إنتاج الأجسام المضادة وعبور الحاجز الدموي الدماغي (BBB)
في المحيط، تتمايز الخلايا البائية إلى أرومات بلازمية تفرز AQP4-IgG. يعزز IL-6 هذا التمايز ويزيد من نفاذية الحاجز الدموي الدماغي. المنطقة الأخيرة هي منطقة تفتقر إلى الحاجز الدموي الدماغي، ويمكن أن تكون طريقًا لدخول AQP4-IgG إلى الجهاز العصبي المركزي.
سلسلة تلف الخلايا النجمية
يرتبط AQP4-IgG بقنوات الماء AQP4 المعبر عنها بكثافة على الأقدام النجمية، مما يؤدي إلى تلف الخلايا النجمية عبر المسارات التالية.
تنشيط المسار الكلاسيكي للمتممة: الجزء Fc من AQP4-IgG ينشط المتممة، مكونًا معقد الهجوم الغشائي (MAC) الذي يتلف الخلايا النجمية مباشرة
السمية الخلوية المعتمدة على الأجسام المضادة (ADCC): تتلف الخلايا القاتلة الطبيعية والعدلات الخلايا النجمية عبر مستقبلات Fcγ
إطلاق C5a أنافيلاتوكسين: يجذب الخلايا المحببة (العدلات والحمضات) مسببًا تلفًا محوريًا ثانويًا وإزالة الميالين1)
في التصلب المتعدد، يكون إزالة الميالين في المادة البيضاء بوساطة الخلايا التائية CD8+ هو السائد، بينما في NMOSD، يكون دور الخلايا التائية CD4+ أكبر، وتتشكل آفات نخرية تشمل كلاً من المادة الرمادية والبيضاء.
سبب التوزع
تتوزع قنوات AQP4 بكثرة في العصب البصري والمنطقة الأخيرة والحبل الشوكي، مما يجعل هذه المناطق أهدافًا انتقائية.
المؤشرات الحيوية
GFAP في المصل: يعكس تلف الخلايا النجمية ويرتفع أثناء النوبات
السلسلة الخفيفة من النيوروفيلمنت في المصل (NfL): يعكس تلف المحاور ويرتبط بشدة النوبة1)
تم الإبلاغ عن السيتوكينات المشاركة مثل IL-6 وIL-10 وIL-17a وG-CSF وTNF-α وBAFF/APRIL.
7. أحدث الأبحاث والتوجهات المستقبلية (تقارير المرحلة البحثية)
يُقدر أن 3-5% من حالات NMOSD تكون مصاحبة للأورام. تمت دراسة الحالات المرتبطة بالورم المسخي المبيضي بشكل خاص بالتفصيل.
أجرى Ikeguchi وآخرون (2021) مراجعة لـ 6 حالات من NMOSD المرتبط بالورم المسخي المبيضي والمصحوب بأجسام مضادة لـ AQP42). جميع الحالات كانت لنساء، متوسط عمر البدء 32.7 سنة (15-50 سنة). من بين 6 حالات، 83% (5/6) عانوا من الغثيان والقيء، و83% كانت لديهم نتائج إيجابية للنطاقات قليلة النسيلة في السائل الدماغي الشوكي، و83% لديهم آفات في جذع الدماغ الظهري. أظهر التحليل المرضي وجود تفاعل مناعي لـ AQP4 وارتشاح لمفاوي في الأنسجة العصبية الإيجابية لـ GFAP داخل الورم، مما يشير إلى آلية يسبب فيها عرض مستضد AQP4 داخل الورم استجابة مناعية ذاتية. بعد استئصال الورم، تحولت الأجسام المضادة لـ AQP4-IgG إلى سلبية في 60% (3/5) من الحالات.
أجرى Ding وآخرون (2021) مراجعة لـ 43 حالة من NMOSD المصاحب للأورام3). 88.4% من الحالات كانت لنساء، وكان سرطان الثدي وسرطان الرئة أكثر أنواع الأورام شيوعًا. تم التأكيد على أهمية فحص الأورام خاصة في مرضى NMOSD الذين تزيد أعمارهم عن 50 عامًا.
يوصى بفحص الأورام بما في ذلك الأورام المسخية حتى في الحالات الشابة.
تم الإبلاغ عن فعالية علاج الامتصاص المناعي بالبروتين A (IA) في حالات NMOSD المقاومة للعلاج والتي لا تستجيب للستيرويدات أو تبادل البلازما أو ريتوكسيماب.
أجرى Fan وآخرون (2024) 3 جلسات من علاج الامتصاص المناعي بالبروتين A لامرأة تبلغ من العمر 35 عامًا مصابة بـ NMOSD المصاحب لمتلازمة شوغرن المقاومة للعلاج والتي لم تستجب للنبضات الستيرويدية والغلوبولين المناعي الوريدي4). في غضون أسبوع واحد، تحسنت بشكل ملحوظ اضطرابات الرؤية والشلل النصفي السفلي واضطرابات الحساسية العميقة، ولوحظ انخفاض سريع في الأجسام المضادة AQP4-IgG و IgA و IgG و IgM. لم يلاحظ أي انتكاس أو تقدم خلال 4 سنوات من المتابعة.
بدأت تتضح صورة انتشار الأمراض المناعية الذاتية المختلفة المصاحبة لـ NMOSD.
أبلغ Zhu وآخرون (2025) عن حالة فتاة تبلغ من العمر 14 عامًا أصيبت بـ NMOSD في سن 11 عامًا5). كانت إيجابية الأجسام المضادة لـ AQP4-IgG، وتم تأكيد إصابتها بمتلازمة شوغرن الأولية خلال المتابعة. تم الحفاظ على الهدأة باستخدام ميثيل بريدنيزولون والغلوبولين المناعي الوريدي وميكوفينولات موفيتيل (MMF) ثم التحول إلى تاكروليموس. تم الإبلاغ عن إصابة 20-30% من حالات NMOSD لدى البالغين بأمراض مناعية ذاتية، ولكن هذه الحالة أظهرت وجود هذه المصاحبة أيضًا في الأطفال5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, Suzuki M, Shimoji K, Motohashi T, Yamamoto T, Shibata N, Kitagawa K. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045. doi:10.1212/NXI.0000000000001045. PMID: 34285095. PMCID: PMC8293286.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain and behavior. 2021;11(10):e2282. doi:10.1002/brb3.2282. PMID:34520629; PMCID:PMC8553315.
Fan W, Chen X, Xiao P, Wei B, Zhang Y, Huang J, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Frontiers in immunology. 2024;15:1429405. doi:10.3389/fimmu.2024.1429405. PMID:39055718; PMCID:PMC11269126.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
انسخ نص المقال والصقه في مساعد الذكاء الاصطناعي الذي تفضله.
تم نسخ المقال إلى الحافظة
افتح أحد مساعدي الذكاء الاصطناعي أدناه والصق النص المنسوخ في مربع المحادثة.