O Transtorno do Espectro da Neuromielite Óptica (NMOSD) é uma doença inflamatória, mediada por anticorpos e autoimune que afeta o sistema nervoso central. Anteriormente conhecida como “doença de Devic”, foi considerada por muitos anos um subtipo da esclerose múltipla (EM). No entanto, com a descoberta dos autoanticorpos contra a aquaporina-4 (AQP4-IgG) em 2004, estabeleceu-se como uma entidade patológica independente.
Contexto histórico: Em 1870, Sir Thomas Clifford Allbutt descreveu pela primeira vez a associação entre mielite e distúrbios do nervo óptico. Pesquisas posteriores mostraram que se trata de uma doença diferente da EM, e hoje é compreendida sob o conceito abrangente de “NMOSD”.
Epidemiologia é a seguinte:
Incidência: A incidência anual estimada de AQP4+NMOSD é de 0,4 a 7,3 por milhão de pessoas1)
Diferença de gênero: A proporção entre os sexos é de aproximadamente 1:9, com clara predominância feminina.
Idade de início: Ocorre principalmente em meia-idade (40–60 anos), com pico no final dos 30 anos ao início dos 40.
Raça: Mais comum em pessoas de ascendência africana e asiática1)
Relação com a gravidez: Cerca de 20–47% das mulheres apresentam o primeiro ataque durante a gravidez ou dentro de um ano após o parto ou aborto.
QQual a diferença entre NMOSD e esclerose múltipla (EM)?
A
NMOSD é uma doença mediada por anticorpos que tem como alvo os canais de água AQP4 nos astrócitos, e difere fundamentalmente da EM em fisiopatologia, tratamento e prognóstico. Na NMOSD, LETM e neurite óptica grave são típicos, e outra diferença importante é que medicamentos modificadores da doença eficazes na EM, como o interferon beta, podem desencadear recaídas na NMOSD.
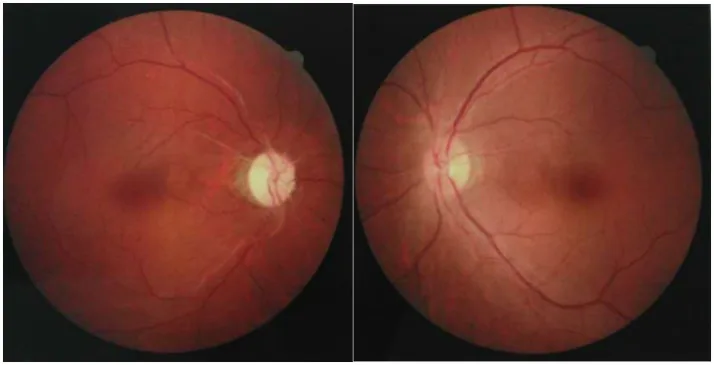
Chuai Y, et al. Paraneoplastic neuromyelitis optica spectrum disorder with dual AQP4-IgG and CRMP5 antibodies following thymectomy: a case report. Front Neurosci. 2026. Figure 1. PMCID: PMC13006573. License: CC BY.
Fotografias de fundo de olho de ambos os olhos, mostrando atrofia óptica com palidez do disco óptico em um olho e leve inchaço do disco óptico no outro olho. Essas imagens retratam os achados do nervo óptico nas fases aguda a crônica da neurite óptica na NMOSD.
Os sintomas da NMOSD variam dependendo da área afetada.
Queda aguda da visão: Um dos principais sintomas. Caracteriza-se por resistência à terapia com esteroides.
Dor ocular: Ocorre com neurite óptica em cerca de metade dos casos.
Anormalidade na visão de cores: Diminuição da saturação do vermelho é típica.
Defeitos de campo visual: Não se limitam a escotoma central, mas podem incluir hemianopsia horizontal, hemianopsia bitemporal e hemianopsia homônima. Isso ocorre porque a lesão se estende ao quiasma óptico e ao trato óptico.
Distúrbios sensoriais e paraplegia: Distúrbios motores e sensoriais devido à mielite.
Distúrbio vesical e retal: Disfunção autonômica associada à mielite
Soluço refratário, náuseas e vômitos: Sintomas característicos devido à lesão da área postrema
Distúrbio da motilidade ocular: Devido a lesão do tronco encefálico
Hipersonia (semelhante à narcolepsia): Devido a lesão diencefálica/hipotalâmica
No início da doença, podem ocorrer sintomas gripais (febre, mialgia, cefaleia).
Bilateral simultâneo: A neurite óptica na NMOSD é bilateral simultânea em 17-82% dos casos. Diferença importante da EM.
Perda visual grave: Na AQP4+NMOSD, a mediana da pior acuidade visual é nível de movimento de mãos (HM). Após recuperação, a mediana permanece em conta-dedos, e 60-69% mantêm deficiência visual permanente de pelo menos 20/200 em um olho.
Mielite
LETM (Mielite Transversa Longitudinal Extensa): Lesão contínua estendendo-se por ≥3 vértebras. Cerca de 85% dos AQP4+NMOSD apresentam isso durante mielite aguda.
Síndrome medular completa: Envolvimento das três vias motora, sensitiva e autonômica.
Disfunção funcional grave: Mais de 30% dos pacientes dependem de cadeira de rodas no pior momento da crise. 37-44% dos AQP4+NMOSD eventualmente necessitam de auxílio para locomoção.
Síndrome da Área Postrema
Soluços refratários: Persistem por dias a semanas e não respondem a antieméticos comuns.
Náuseas e vômitos: Como a área postrema carece de barreira hematoencefálica, é um local de fácil acesso direto para AQP4-IgG.
Achados diagnósticos centrais da NMOSD: Soluços refratários inexplicáveis são um gatilho para suspeitar ativamente de NMOSD.
QComo a neurite óptica na NMOSD difere da relacionada à EM?
A
A neurite óptica na NMOSD é mais grave, bilateral e recorrente, com pior prognóstico visual. Na NMOSD AQP4-positiva, estima-se que 60-69% dos pacientes apresentem deficiência visual permanente de pelo menos 20/200 em um olho. Além disso, tende a envolver o quiasma óptico, causando diversos defeitos de campo visual, como hemianopsia bitemporal, o que a diferencia da EM.
RM da Medula Espinhal: LETM é o mais característico. Predomínio da substância cinzenta central. Acompanhado de edema medular, hipossinal em T1 e realce pelo Gd. Cerca de 85% dos pacientes com NMOSD AQP4+ apresentam LETM durante mielite aguda 1)
RM do Nervo Óptico: Sequência de supressão de gordura é obrigatória. Inflamação bilateral e de longo segmento (>50%) é característica. Envolvimento da porção posterior e do quiasma óptico é típico de NMOSD AQP4+ 1)
RM do Encéfalo: Lesões da área postrema, lesões do tronco encefálico periventriculares do quarto ventrículo, lesões hipotalâmicas/periventriculares do terceiro ventrículo e lesões extensas da substância branca.
Características da NMOSD: Diferente da EM, novas lesões T2 assintomáticas são raras (3-13%). RM de vigilância geralmente não é necessária 1)
Em idosos, é importante diferenciar de neuropatia óptica isquêmica, mielopatia cervical, infarto medular e linfoma primário do SNC.
QO NMOSD pode ser diagnosticado mesmo se o anticorpo AQP4 for negativo?
A
Sim. Mesmo com AQP4-IgG negativo ou não testado, se dois ou mais achados clínicos principais forem atendidos, os requisitos adicionais de RM forem cumpridos e outras doenças forem excluídas, o NMOSD pode ser diagnosticado. Além disso, cerca de 30% dos casos AQP4-IgG negativos são positivos para MOG-IgG, e a dosagem de ambos os anticorpos é recomendada. Menos de 1% dos casos AQP4-IgG negativos sofrem soroconversão posteriormente.
Primeira escolha: Terapia com pulsos de esteroides
Metilprednisolona 1.000 mg/dia por via intravenosa por 3 dias
Se não houver melhora da visão, considerar repetir 1 ciclo após intervalo de 3-4 dias
A neurite óptica na NMOSD é altamente resistente a esteroides, portanto, se a resposta for insuficiente, considerar o próximo tratamento precocemente
Segunda escolha: Terapia de troca plasmática
Realizada quando não há resposta aos pulsos de esteroides. As seguintes opções estão disponíveis:
Troca plasmática simples (PE): Maior eficácia, mas também maior dano ao organismo
Troca plasmática por filtração de membrana dupla (DFPP)
Terapia de imunoadsorção (IA): Remoção seletiva de anticorpos
A ordem de eficácia é considerada: troca plasmática simples > filtração de membrana dupla > imunoadsorção. Realiza-se 5-6 sessões por ciclo, e após o tratamento é necessária internação até a recuperação dos níveis de IgG. Note que para “neurite óptica” pode não ser coberto pelo seguro, exigindo explicação ao paciente.
Na AQP4+NMOSD, a terapia de manutenção deve ser iniciada precocemente após o primeiro ataque 1). Após a troca plasmática, é comum a transição para prednisolona 5-10 mg/dia + azatioprina 50-100 mg/dia.
Agentes biológicos com alto nível de evidência são os seguintes:
Inibidores do complemento
Eculizumabe: 900 mg IV semanalmente × 4 vezes → 1.200 mg a cada 2 semanas como dose de manutenção 1)
Ravulizumabe: Dose de ataque baseada no peso (2.400–3.000 mg) → após o dia 15, 3.000–3.600 mg a cada 8 semanas1)
Terapia de depleção de células B
Rituximabe: 375 mg/m² IV semanalmente × 4 vezes, ou 1.000 mg × 2 (com intervalo de 2 semanas) → 1.000 mg × 2 a cada 6 meses1)
Inebilizumabe: 300 mg IV a cada 15 dias por 2 vezes → a cada 6 meses1)
Inibidores do receptor de IL-6
Satralizumabe: 120 mg subcutâneo, a cada 4 semanas1)
QO que fazer se a pulsoterapia com esteroides não for eficaz?
A
A plasmaférese é a próxima opção. Existem três tipos: plasmaférese simples, plasmaférese por filtração de membrana dupla e imunoadsorção. A plasmaférese simples é considerada a mais eficaz, mas também a mais desgastante para o organismo. Um ciclo consiste em 5 a 6 sessões, e o tratamento requer internação hospitalar.
A NMOSD é essencialmente uma astrocitopatia. O mecanismo da doença é o seguinte:
Produção de anticorpos e passagem pela barreira hematoencefálica (BHE)
Na periferia, células B se diferenciam em plasmablastos secretores de AQP4-IgG. A IL-6 facilita essa diferenciação e aumenta a permeabilidade da barreira hematoencefálica. A área postrema é uma região que carece de barreira hematoencefálica e pode ser uma via de entrada do AQP4-IgG no SNC.
Cascata de lesão astrocitária
O AQP4-IgG se liga aos canais de água AQP4 altamente expressos nos pés dos astrócitos, causando lesão astrocitária pelas seguintes vias.
Ativação da via clássica do complemento: A porção Fc do AQP4-IgG ativa o complemento, formando o complexo de ataque à membrana (MAC) que lesa diretamente os astrócitos
ADCC (citotoxicidade dependente de anticorpo): Células NK e neutrófilos lesam astrócitos via receptores Fcγ
Liberação de C5a anafilatoxina: Recruta granulócitos (neutrófilos, eosinófilos) causando dano axonal secundário e desmielinização1)
Diferenças patológicas com a EM
Na EM, a desmielinização centrada na substância branca mediada por células T CD8+ é predominante, enquanto na NMOSD, o envolvimento de células T CD4+ é maior, formando lesões necróticas que afetam tanto a substância cinzenta quanto a branca.
Razão da distribuição
Os canais AQP4 são abundantemente distribuídos no nervo óptico, área postrema e medula espinhal, tornando essas áreas alvos seletivos.
Biomarcadores
GFAP sérico: Reflete lesão astrocitária e eleva-se durante as crises
Neurofilamento de cadeia leve sérico (NfL): Reflete dano axonal e correlaciona-se com a gravidade da crise1)
As citocinas envolvidas incluem IL-6, IL-10, IL-17a, G-CSF, TNF-α e BAFF/APRIL foram relatadas.
7. Pesquisas mais recentes e perspectivas futuras (relatórios em fase de pesquisa)
Estima-se que 3-5% das NMOSD sejam paraneoplásicas. Casos associados a teratoma ovariano foram estudados em detalhes.
Ikeguchi et al. (2021) realizaram uma revisão de 6 casos de NMOSD associada a teratoma ovariano com AQP4-positivo2). Todos os pacientes eram mulheres, idade média de início 32,7 anos (15-50 anos). Dos 6 casos, 83% (5/6) apresentaram náuseas e vômitos, 83% tiveram bandas oligoclonais no LCR positivas e 83% apresentaram lesões no tronco encefálico dorsal. A análise patológica confirmou reatividade imune AQP4 e infiltração linfocítica no tecido neural GFAP-positivo dentro do tumor, sugerindo um mecanismo no qual a apresentação do antígeno AQP4 dentro do tumor desencadeia uma resposta autoimune. Após a ressecção do tumor, o AQP4-IgG tornou-se negativo em 60% (3/5) dos casos.
Ding et al. (2021) realizaram uma revisão de 43 casos de NMOSD paraneoplásica3). 88,4% eram mulheres, com câncer de mama e pulmão sendo os tipos tumorais mais comuns. A importância do rastreamento de tumores, especialmente em pacientes com NMOSD com mais de 50 anos, foi enfatizada.
O rastreamento de tumores, incluindo teratoma, é recomendado mesmo em casos jovens.
A eficácia da terapia de imunoadsorção com Proteína-A (IA) foi relatada em NMOSD refratária que não responde a esteroides, plasmaférese ou rituximabe.
Fan et al. (2024) realizaram 3 sessões de imunoadsorção com Proteína-A em uma mulher de 35 anos com NMOSD associada à síndrome de Sjögren refratária que não respondeu a pulsoterapia com esteroides e IVIG4). Dentro de uma semana, os distúrbios visuais, paraplegia e distúrbios proprioceptivos melhoraram significativamente, e foi observada uma rápida diminuição de AQP4-IgG, IgA, IgG e IgM. Nenhuma recaída ou progressão foi observada durante 4 anos de acompanhamento.
Características dos casos com comorbidade de doenças autoimunes
O panorama real de várias doenças autoimunes que acompanham a NMOSD está se tornando claro.
Zhu et al. (2025) relataram o caso de uma menina de 14 anos que desenvolveu NMOSD aos 11 anos de idade5). AQP4-IgG positivo, e a síndrome de Sjögren primária foi confirmada durante o curso. A remissão foi mantida com metilprednisolona, IVIG e micofenolato de mofetila (MMF) seguido de mudança para tacrolimo. Comorbidade de doenças autoimunes é relatada em 20-30% dos casos adultos de NMOSD, mas este caso mostrou que essa comorbidade também pode ocorrer em crianças5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, Suzuki M, Shimoji K, Motohashi T, Yamamoto T, Shibata N, Kitagawa K. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045. doi:10.1212/NXI.0000000000001045. PMID: 34285095. PMCID: PMC8293286.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain and behavior. 2021;11(10):e2282. doi:10.1002/brb3.2282. PMID:34520629; PMCID:PMC8553315.
Fan W, Chen X, Xiao P, Wei B, Zhang Y, Huang J, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Frontiers in immunology. 2024;15:1429405. doi:10.3389/fimmu.2024.1429405. PMID:39055718; PMCID:PMC11269126.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.