Nöromiyelitis Optika Spektrum Bozukluğu (NMOSD), merkezi sinir sistemini etkileyen inflamatuvar, antikor aracılı, otoimmün bir hastalıktır. Eskiden “Devic hastalığı” olarak da adlandırılmış ve uzun yıllar multipl sklerozun (MS) bir alt tipi olarak kabul edilmiştir. Ancak 2004 yılında aquaporin-4’e (AQP4) karşı otoantikorların (AQP4-IgG) keşfiyle bağımsız bir hastalık birimi olarak yerleşmiştir.
Tarihsel arka plan: 1870’te Sir Thomas Clifford Allbutt, miyelit ile optik sinir bozukluğu arasındaki ilişkiyi ilk kez tanımlamıştır. Sonraki araştırmalar, bunun MS’ten farklı bir hastalık olduğunu ortaya koymuş ve günümüzde “NMOSD” kapsayıcı kavramı altında ele alınmaktadır.
Epidemiyoloji aşağıdaki gibidir:
İnsidans: AQP4+ NMOSD’nin tahmini yıllık insidansı milyonda 0,4 ila 7,3’tür1)
Cinsiyet farkı: Kadın/erkek oranı yaklaşık 1:9 olup kadınlarda belirgin bir baskınlık vardır.
Başlangıç yaşı: Çoğunlukla orta yaşta (40-60 yaş) görülür. 30’lu yaşların sonu ile 40’lı yaşların başında pik yapar
Irk: Afrikalı ve Asyalılarda daha sık görülme eğilimi vardır1)
Gebelikle ilişkisi: Kadınların yaklaşık %20-47’si gebelik sırasında veya doğum/düşükten sonraki bir yıl içinde ilk atağı geçirir
QNMOSD ile multipl skleroz (MS) arasındaki fark nedir?
A
NMOSD, astrositlerdeki AQP4 su kanalını hedef alan antikor aracılı bir hastalıktır ve patofizyoloji, tedavi ve prognoz açısından MS’ten temel olarak farklıdır. NMOSD’de LETM ve şiddetli optik nörit tipiktir ve MS’te etkili olan interferon beta gibi hastalık modifiye edici ilaçların NMOSD’de atakları tetikleyebilmesi önemli bir farktır.
Chuai Y, et al. Paraneoplastic neuromyelitis optica spectrum disorder with dual AQP4-IgG and CRMP5 antibodies following thymectomy: a case report. Front Neurosci. 2026. Figure 1. PMCID: PMC13006573. License: CC BY.
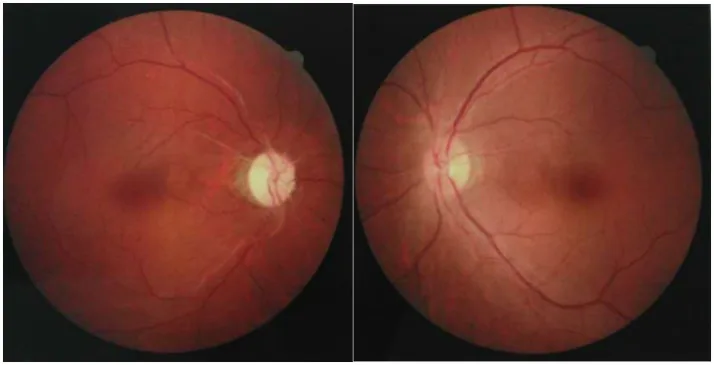
Her iki gözün fundus fotoğraflarında, bir gözde optik disk solukluğuna bağlı optik atrofi, diğer gözde hafif optik disk şişmesi görülmektedir. NMOSD’de görülen optik nöritin akut ve kronik dönem optik sinir bulguları gösterilmiştir.
NMOSD’nin belirtileri etkilenen bölgeye göre değişir.
Ani görme azalması: Ana belirtilerden biridir. Steroid tedavisine direnç karakteristiktir
Göz ağrısı: Optik nörite eşlik eder, vakaların yaklaşık yarısında görülür
Renk görme bozukluğu: Kırmızı renk doygunluğunda azalma tipiktir
Görme alanı defekti: Santral skotomla sınırlı kalmayıp horizontal hemianopsi, bitemporal hemianopsi ve homonim hemianopsi de görülebilir. Bunun nedeni lezyonun kiazma ve optik traktusa kadar uzanmasıdır.
Duyusal bozukluklar ve parapleji: Miyelite bağlı motor ve duyusal bozukluklar
Mesane-barsak disfonksiyonu: Miyelite bağlı otonomik bozukluk
İnatçı hıçkırık ve bulantı/kusma: Area postrema lezyonuna bağlı karakteristik semptomlar
Göz hareket bozuklukları: Beyin sapı lezyonuna bağlı
Aşırı uyku hali (narkolepsi benzeri): Diensefalon/hipotalamus lezyonuna bağlı
Hastalığın başlangıcında grip benzeri semptomlar (ateş, kas ağrısı, baş ağrısı) görülebilir.
Ciddi görme kaybı: AQP4+NMOSD’de en kötü görme keskinliği medyanı el hareketi (HM) düzeyindedir. İyileşme sonrası medyan parmak sayma düzeyinde kalır ve hastaların %60-69’u en az bir gözde 20/200 veya daha kötü kalıcı görme bozukluğu yaşar.
Miyelit
LETM (Uzun segment transvers miyelit): Üç veya daha fazla vertebrayı kapsayan sürekli lezyon. AQP4+NMOSD’li hastaların yaklaşık %85’i akut miyelit sırasında bu paterni gösterir.
Komplet spinal sendrom: Motor, duyusal ve otonomik yolların tümünün tutulumu.
Ciddi fonksiyonel kayıp: Hastaların %30’undan fazlası atak sırasında tekerlekli sandalyeye bağımlı hale gelir. AQP4+NMOSD’li hastaların %37-44’ü sonunda yürüme yardımcısına ihtiyaç duyar.
Area postrema sendromu
Tedaviye dirençli hıçkırık : Günler ila haftalar sürer ve normal antiemetiklere yanıt vermez.
Bulantı ve kusma : Area postrema kan-beyin bariyerinden yoksun olduğu için AQP4-IgG’nin doğrudan ulaşması kolaydır.
NMOSD tanısında temel bulgu : Açıklanamayan tedaviye dirençli hıçkırık, NMOSD’den aktif olarak şüphelenmek için bir nedendir.
QNMOSD'deki optik nörit, MS ile ilişkili olandan nasıl farklıdır?
A
NMOSD’de optik nörit daha şiddetli, bilateral ve tekrarlayıcıdır ve görme prognozu kötüdür. AQP4+ NMOSD’de hastaların %60-69’unda en az bir gözde 20/200 veya daha kötü kalıcı görme kaybı gelişir. Ayrıca kiazma tutulumu daha sıktır ve bitemporal hemianopsi gibi çeşitli görme alanı defektlerine yol açar; bu da MS’ten farklıdır.
Spinal MRG: LETM en karakteristik bulgudur. Santral gri cevher baskındır. Spinal kord şişliği, T1 hipointensitesi ve Gd kontrastlanması eşlik eder. AQP4+ NMOSD’li hastaların yaklaşık %85’i akut miyelit sırasında LETM gösterir1)
Optik sinir MRG: Yağ baskılamalı görüntüler zorunludur. Bilateral ve uzun segment inflamasyon (%50’den fazla) karakteristiktir. Posterior kısım ve kiazma tutulumu AQP4+ NMOSD için tipiktir1)
Beyin MRG: Area postrema lezyonları, dördüncü ventrikül çevresi beyin sapı lezyonları, hipotalamus/üçüncü ventrikül çevresi lezyonları ve yaygın beyaz cevher lezyonları görülür
NMOSD özellikleri: MS’ten farklı olarak, asemptomatik yeni T2 lezyonları nadirdir (%3-13). Sürveyans MRG genellikle gerekli değildir1)
Yaşlılarda iskemik optik nöropati, servikal spondilotik miyelopati, spinal kord enfarktüsü ve primer CNS lenfoması ile ayırıcı tanı önemlidir
QAQP4 antikoru negatif olsa bile NMOSD tanısı konulabilir mi?
A
Evet. AQP4-IgG negatif veya test edilmemiş olsa bile, en az iki ana klinik özellik karşılanırsa, ek MRG kriterleri karşılanırsa ve diğer hastalıklar dışlanırsa NMOSD tanısı konulabilir. Ayrıca, AQP4-IgG negatif vakaların yaklaşık %30’unda MOG-IgG pozitiftir ve her iki antikorun ölçümü önerilir. AQP4-IgG negatif vakaların %1’inden azı daha sonra serokonversiyon gösterir.
Metilprednizolon 1000 mg/gün intravenöz infüzyon 3 gün süreyle uygulanır
Görme düzelmezse 3-4 gün ara verilerek tekrar bir kür düşünülür
NMOSD’de optik nörit steroide dirençli olduğundan, yanıt yetersizse erken dönemde sonraki tedavi düşünülmelidir
İkinci basamak: Plazma değişimi tedavisi
Steroid pulse’a yanıt vermeyen durumlarda uygulanır. Aşağıdaki seçenekler mevcuttur:
Basit plazma değişimi (PE): En etkili ancak vücuda en fazla hasar veren
Çift membran filtrasyon plazma değişimi (DFPP)
İmmün adsorpsiyon (IA): Antikorların seçici olarak uzaklaştırılması mümkün
Etkinlik sırası: basit plazma değişimi > çift membran filtrasyon > immün adsorpsiyon. Bir kür 5-6 seans uygulanır ve tedavi sonrası vücut IgG düzeyi düzelene kadar hastanede yatış gerekir. “Optik nörit” için sigorta kapsamı dışında olabilir, hastaya açıklama yapılmalıdır.
AQP4+ NMOSD’de ilk ataktan itibaren idame tedavisine erken başlanması önerilir 1). Plazma değişimi sonrası prednizolon 5-10 mg/gün + azatioprin 50-100 mg/gün’e geçiş yaygındır.
Kanıt düzeyi yüksek biyolojik ajanlar şunlardır:
Kompleman inhibitörleri
Ekulizumab: 900 mg IV haftada bir × 4 kez, ardından 1200 mg iki haftada bir idame dozu 1)
Ravilizumab: Vücut ağırlığına dayalı yükleme dozu (2400-3000 mg) → 15. günden itibaren 3000-3600 mg her 8 haftada bir 1)
B hücre yok edici tedavi
Rituksimab: 375 mg/m² IV haftada bir × 4 kez veya 1000 mg × 2 kez (2 hafta arayla) → ardından 6 ayda bir 1000 mg × 2 kez 1)
İnebliZumab: 300 mg IV 15 günde bir 2 kez → ardından 6 ayda bir 1)
IL-6 reseptör inhibitörleri
Satralizumab: 120 mg subkutan, 4 haftada bir 1)
QSteroid puls tedavisi işe yaramazsa ne yapılmalıdır?
A
Plazma değişimi bir sonraki seçenektir. Basit plazma değişimi, çift membran filtrasyon plazma değişimi ve immün adsorpsiyon olmak üzere üç tür arasından seçim yapılır. Basit plazma değişimi en etkili olarak kabul edilir ancak vücut üzerinde daha fazla yük oluşturur. Bir kür 5-6 seans uygulanır ve tedavi sonrası hastanede yatış gerekir.
6. Patofizyoloji ve ayrıntılı hastalık mekanizması
NMOSD temelde bir astrosit hastalığıdır (astrositopati). Hastalık mekanizması şu şekildedir:
Antikor üretimi ve kan-beyin bariyerini (KBB) geçiş
Periferde B hücreleri, AQP4-IgG salgılayan plazmablastlara farklılaşır. IL-6 bu farklılaşmayı hızlandırır ve kan-beyin bariyerinin geçirgenliğini artırır. Area postrema, kan-beyin bariyeri olmayan bir bölgedir ve AQP4-IgG’nin CNS’ye girişi için bir yol olabilir.
Astrosit hasar zinciri
AQP4-IgG, astrosit ayak uçlarında yoğun olarak eksprese edilen AQP4 su kanallarına bağlanır ve aşağıdaki yollarla astrosit hasarına neden olur.
Klasik kompleman yolunun aktivasyonu: AQP4-IgG’nin Fc kısmı komplemanı aktive eder ve membran atak kompleksi (MAC) oluşturarak astrositlere doğrudan zarar verir.
ADCC (Antikor bağımlı hücresel sitotoksisite): NK hücreleri ve nötrofiller, Fcγ reseptörleri aracılığıyla astrositlere zarar verir.
C5a anafilatoksin salınımı: Granülositleri (nötrofil ve eozinofil) çekerek sekonder akson hasarı ve demiyelinizasyona neden olur1).
MS’de, CD8+ T hücrelerinin merkezi rol oynadığı beyaz cevher ağırlıklı demiyelinizasyon baskınken, NMOSD’de CD4+ T hücrelerinin katkısı daha fazladır ve hem gri hem de beyaz cevheri içeren nekrotik lezyonlar oluşur.
Dağılımın nedeni
AQP4 kanalları optik sinir, area postrema ve omurilikte bol miktarda bulunur ve bu bölgeler seçici olarak hedef alınır.
Biyobelirteçler
Serum GFAP: Astrosit hasarını yansıtır ve atak sırasında yükselir.
Serum nörofilament hafif zinciri (NfL): Akson hasarını yansıtır ve atak şiddeti ile korelasyon gösterir1).
İlgili sitokinler olarak IL-6, IL-10, IL-17a, G-CSF, TNF-α ve BAFF/APRIL rapor edilmiştir.
7. En yeni araştırmalar ve geleceğe bakış (araştırma aşamasındaki raporlar)
NMOSD vakalarının %3-5’inin paraneoplastik olduğu tahmin edilmektedir. Over teratomu ile ilişkili vakalar özellikle ayrıntılı olarak incelenmiştir.
Ikeguchi ve ark. (2021), over teratomu ile ilişkili AQP4+ NMOSD’li 6 vakanın incelemesini gerçekleştirdi2). Tümü kadın, ortalama başlangıç yaşı 32.7 (15-50). 6 vakanın %83’ünde (5/6) bulantı ve kusma görüldü, %83’ünde BOS oligoklonal bandı pozitifti, %83’ünde dorsal beyin sapı lezyonu vardı. Patolojik incelemede, tümör içindeki GFAP pozitif sinir dokusunda AQP4 immünreaktivitesi ve lenfosit infiltrasyonu doğrulandı ve tümör içinde AQP4 antijen sunumunun otoimmün yanıtı tetiklediği bir mekanizma öne sürüldü. Tümör çıkarıldıktan sonra vakaların %60’ında (3/5) AQP4-IgG negatifleşti.
Ding ve ark. (2021), 43 paraneoplastik NMOSD vakasının incelemesini gerçekleştirdi3). %88.4’ü kadındı ve meme kanseri ile akciğer kanseri en sık görülen tümör tipleriydi. Özellikle 50 yaş üstü NMOSD hastalarında tümör taramasının önemi vurgulanmıştır.
Genç vakalarda bile teratom dahil tümör taraması önerilir.
Steroid, plazma değişimi ve rituksimab gibi tedavilere yanıt vermeyen dirençli NMOSD için Protein-A immünadsorpsiyon (IA) tedavisinin etkinliği rapor edilmiştir.
Fan ve ark. (2024), steroid pulse ve IVIG’ye yanıt vermeyen dirençli Sjögren sendromu ile birlikte NMOSD’li 35 yaşında bir kadın hastaya 3 seans Protein-A immünadsorpsiyon uyguladı4). Bir hafta içinde görme bozukluğu, parapleji ve propriyoseptif duyu bozukluğu belirgin şekilde düzeldi ve AQP4-IgG, IgA, IgG ve IgM’de hızlı düşüş doğrulandı. 4 yıllık takipte nüks veya ilerleme gözlenmedi.
Otoimmün hastalıklarla birlikte görülen vakaların özellikleri
NMOSD’ye çeşitli otoimmün hastalıkların eşlik etme durumu giderek netleşmektedir.
Zhu ve ark. (2025), 11 yaşında NMOSD gelişen 14 yaşında bir kız çocuğu vakasını bildirdi5). AQP4-IgG pozitifti ve seyir sırasında primer Sjögren sendromu birlikteliği doğrulandı. Metilprednizolon, IVIG ve mikofenolat mofetil (MMF)‘den takrolimusa geçiş ile remisyon sağlandı. Erişkin NMOSD vakalarının %20-30’unda otoimmün hastalık birlikteliği rapor edilmiştir, ancak bu vaka çocuklarda da birlikteliğin olabileceğini göstermiştir5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, Suzuki M, Shimoji K, Motohashi T, Yamamoto T, Shibata N, Kitagawa K. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045. doi:10.1212/NXI.0000000000001045. PMID: 34285095. PMCID: PMC8293286.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain and behavior. 2021;11(10):e2282. doi:10.1002/brb3.2282. PMID:34520629; PMCID:PMC8553315.
Fan W, Chen X, Xiao P, Wei B, Zhang Y, Huang J, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Frontiers in immunology. 2024;15:1429405. doi:10.3389/fimmu.2024.1429405. PMID:39055718; PMCID:PMC11269126.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.