Il disturbo dello spettro della neuromielite ottica (NMOSD) è una malattia infiammatoria, autoimmunitaria e mediata da anticorpi che colpisce il sistema nervoso centrale. In passato chiamata anche «malattia di Devic», è stata a lungo considerata un sottotipo della sclerosi multipla (SM). Tuttavia, la scoperta nel 2004 degli autoanticorpi contro l’acquaporina-4 (AQP4-IgG) l’ha stabilita come entità patologica indipendente.
Contesto storico: Nel 1870, Sir Thomas Clifford Allbutt descrisse per primo l’associazione tra mielite e disturbi ottici. Ricerche successive hanno riconosciuto che si tratta di una malattia diversa dalla SM, e ora è compresa nel concetto globale di «NMOSD».
Epidemiologia: Quanto segue.
Incidenza: L’incidenza annuale stimata del NMOSD con anticorpi anti-AQP4 è di 0,4-7,3 per milione di abitanti1)
Rapporto tra i sessi: Il rapporto maschi:femmine è di circa 1:9, con una netta predominanza femminile.
Età di insorgenza: prevalentemente in età media (40-60 anni). Picco tra la fine dei 30 e l’inizio dei 40 anni.
Etnia: tendenza a essere più frequente in persone di origine africana e asiatica1)
Relazione con la gravidanza: circa il 20-47% delle donne presenta il primo episodio durante la gravidanza o entro un anno dal parto o dall’aborto.
QQual è la differenza tra NMOSD e sclerosi multipla (SM)?
A
La NMOSD è una malattia anticorpo-mediata che colpisce il canale acquoso AQP4 degli astrociti, con fisiopatologia, trattamento e prognosi fondamentalmente diversi dalla SM. Nella NMOSD sono tipici la LETM e la neurite ottica grave, e farmaci modificanti la malattia efficaci nella SM, come l’interferone beta, possono scatenare ricadute di NMOSD, un’importante differenza.
Chuai Y, et al. Paraneoplastic neuromyelitis optica spectrum disorder with dual AQP4-IgG and CRMP5 antibodies following thymectomy: a case report. Front Neurosci. 2026. Figure 1. PMCID: PMC13006573. License: CC BY.
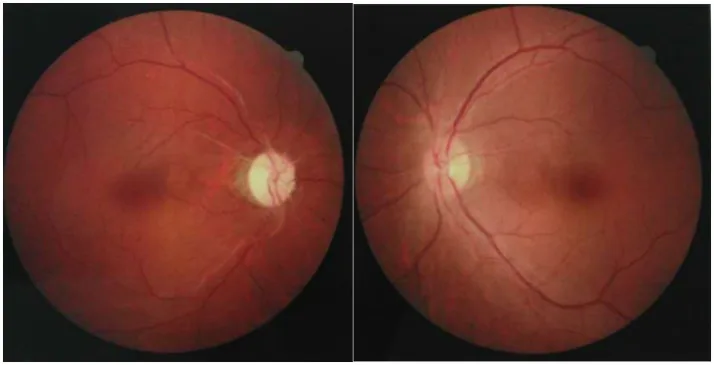
Fotografie del fondo oculare di entrambi gli occhi che mostrano atrofia ottica per pallore del disco in un occhio e lieve gonfiore del disco nell’altro occhio. Sono rappresentati i segni ottici della fase acuta e cronica della neurite ottica osservata nella NMOSD.
I sintomi della NMOSD variano a seconda della sede colpita.
Rapida riduzione dell’acuità visiva: uno dei sintomi principali. Caratterizzata da resistenza al trattamento steroideo.
Dolore oculare: associato a neurite ottica, presente in circa la metà dei casi.
Alterazione della visione dei colori: tipica la riduzione della saturazione del rosso.
Difetti del campo visivo: non limitati a scotoma centrale, ma possono includere emianopsia orizzontale, emianopsia bitemporale o emianopsia omonima, poiché le lesioni si estendono al chiasma e al tratto ottico.
Disturbi sensitivi e paraplegia: disturbi motori e sensitivi dovuti a mielite.
Disturbi vescico-rettali : disfunzione autonomica associata a mielite
Singhiozzo intrattabile e nausea/vomito : sintomi caratteristici da lesione dell’area postrema
Disturbi oculomotori : da lesione del tronco encefalico
Ipersonnia (simile alla narcolessia) : da lesione diencefalica/ipotalamica
All’esordio della malattia possono manifestarsi sintomi simil-influenzali (febbre, mialgie, cefalea).
Bilaterale simultanea : la neurite ottica nella NMOSD è bilaterale simultanea nel 17-82% dei casi. Differenza importante con la SM.
Grave calo visivo : nella NMOSD AQP4+, la mediana dell’acuità visiva minima è a livello di percezione del movimento della mano (HM). Dopo il recupero, la mediana rimane a livello di conteggio delle dita, e il 60-69% dei pazienti presenta un deficit visivo permanente di almeno 20/200 in almeno un occhio.
Mielite
LETM (mielite trasversa longitudinalmente estesa) : lesione continua che si estende per almeno 3 vertebre. Circa l’85% dei pazienti con NMOSD AQP4+ la presenta durante la mielite acuta.
Sindrome spinale completa : coinvolgimento di tutte e tre le vie (motoria, sensitiva, autonomica).
Grave compromissione funzionale : oltre il 30% dei pazienti è dipendente dalla sedia a rotelle al nadir dell’attacco. Il 37-44% dei pazienti con NMOSD AQP4+ necessita infine di ausili per la deambulazione.
Sindrome dell'area postrema
Singhiozzo refrattario : persiste per giorni o settimane e non risponde ai comuni antiemetici.
Nausea e vomito : poiché l’area postrema è priva di barriera emato-encefalica, l’AQP4-IgG può raggiungerla direttamente.
Segno diagnostico chiave della NMOSD : un singhiozzo refrattario inspiegabile deve far sospettare attivamente una NMOSD.
QIn che modo la neurite ottica della NMOSD differisce da quella associata alla SM?
A
La neurite ottica nella NMOSD è più grave, bilaterale e ricorrente, con una prognosi visiva sfavorevole. Nei pazienti con NMOSD AQP4+, il 60-69% presenta un deficit visivo permanente di almeno 20/200 in un occhio. Inoltre, il chiasma ottico è frequentemente coinvolto, causando vari difetti del campo visivo come l’emianopsia bitemporale, a differenza della SM.
RM midollare: la LETM è la più caratteristica. Predominanza della sostanza grigia centrale. Con edema midollare, ipointensità T1 e enhancement dopo Gd. Circa l’85% dei NMOSD AQP4+ presenta LETM durante la mielite acuta 1)
RM dei nervi ottici: le sequenze con soppressione del grasso sono essenziali. Infiammazione bilaterale e a lungo segmento (>50%) è caratteristica. Il coinvolgimento della porzione posteriore e del chiasma è tipico dei NMOSD AQP4+ 1)
RM encefalica: lesioni dell’area postrema, del tronco encefalico periventricolare al IV ventricolo, dell’ipotalamo/regione periventricolare al III ventricolo, estese lesioni della sostanza bianca, ecc.
Caratteristiche dei NMOSD: a differenza della SM, le nuove lesioni T2 asintomatiche sono rare (3-13%). La RM di sorveglianza di solito non è necessaria 1)
Negli anziani è importante differenziare dalla neuropatia ottica ischemica, mielopatia spondilotica cervicale, infarto midollare e linfoma primitivo del SNC
QÈ possibile diagnosticare NMOSD anche se gli anticorpi anti-AQP4 sono negativi?
A
Sì. Anche in caso di AQP4-IgG negativo o non testato, se sono soddisfatte almeno due caratteristiche cliniche principali, i criteri RM aggiuntivi sono soddisfatti e altre malattie sono escluse, si può diagnosticare NMOSD. Inoltre, circa il 30% dei casi AQP4-IgG negativi è positivo per MOG-IgG, e si raccomanda la determinazione di entrambi gli anticorpi. Meno dell’1% dei casi AQP4-IgG negativi sieroconverte successivamente.
Somministrare metilprednisolone 1.000 mg/die per via endovenosa per 3 giorni
Se non si osserva miglioramento dell’acuità visiva, considerare un altro ciclo dopo un intervallo di 3-4 giorni
La neurite ottica nella NMOSD è spesso resistente agli steroidi; in caso di risposta insufficiente, considerare precocemente la terapia successiva
Seconda linea: plasmaferesi
Eseguita in caso di mancata risposta ai boli di steroidi. Le seguenti opzioni sono disponibili:
Plasmaferesi semplice (PE) : la più efficace ma anche la più aggressiva per l’organismo
Plasmaferesi a doppia filtrazione (DFPP)
Immunoadsorbimento (IA) : consente la rimozione selettiva degli anticorpi
L’efficacia è classificata come segue: plasmaferesi semplice > doppia filtrazione > immunoadsorbimento. Un ciclo comprende 5-6 sedute; dopo il trattamento è necessario il ricovero fino al recupero dei livelli di IgG sieriche. Nota: per la neurite ottica, questi trattamenti potrebbero non essere coperti dall’assicurazione, il che richiede una spiegazione al paziente.
Prevenzione delle recidive (terapia di mantenimento)
Nella NMOSD AQP4+, si raccomanda di iniziare precocemente la terapia di mantenimento dopo il primo attacco 1). Dopo la plasmaferesi, di solito si passa a prednisolone 5-10 mg/die + azatioprina 50-100 mg/die.
I farmaci biologici con elevato livello di evidenza sono:
Inibitori del complemento
Eculizumab : 900 mg EV settimanale × 4, poi 1.200 mg ogni 2 settimane come dose di mantenimento 1)
Ravulizumab : dose di carico basata sul peso (2.400–3.000 mg) → dal giorno 15, 3.000–3.600 mg ogni 8 settimane1)
Terapia di deplezione dei linfociti B
Rituximab : 375 mg/m² EV settimanale × 4, oppure 1.000 mg × 2 (a distanza di 2 settimane) → ogni 6 mesi 1.000 mg × 21)
Inebilizumab : 300 mg EV ogni 15 giorni × 2 → ogni 6 mesi1)
Inibitori del recettore dell’IL-6
Satralizumab : 120 mg per via sottocutanea ogni 4 settimane1)
QCosa fare se la terapia con boli di steroidi non è efficace?
A
La plasmaferesi è l’opzione successiva. Si sceglie tra tre tipi: scambio plasmatico semplice, plasmaferesi a doppia filtrazione e immunoassorbimento. Lo scambio plasmatico semplice è considerato il più efficace ma anche il più gravoso per l’organismo. Viene eseguito un ciclo di 5-6 sedute, e dopo il trattamento è necessario il ricovero ospedaliero.
6. Fisiopatologia e meccanismi dettagliati della malattia
La NMOSD è essenzialmente un’astrocitopatia. Il meccanismo della malattia è il seguente:
Produzione di anticorpi e passaggio attraverso la barriera emato-encefalica (BEE)
In periferia, i linfociti B si differenziano in plasmablasti secernenti AQP4-IgG. L’IL-6 promuove questa differenziazione e aumenta la permeabilità della barriera emato-encefalica (BEE). L’area postrema è una regione priva di BEE e può costituire una via di ingresso per le AQP4-IgG nel SNC.
Catena del danno astrocitario
Le AQP4-IgG si legano ai canali idrici AQP4, altamente espressi sui pedicelli astrocitari, causando danno astrocitario attraverso le seguenti vie.
Attivazione della via classica del complemento: La porzione Fc delle AQP4-IgG attiva il complemento, formando il complesso di attacco alla membrana (MAC) che danneggia direttamente gli astrociti.
ADCC (citotossicità cellulare anticorpo-dipendente): Cellule NK e neutrofili danneggiano gli astrociti tramite recettori Fcγ.
Rilascio di C5a anafilotossina: Recluta granulociti (neutrofili, eosinofili), causando danno assonale secondario e demielinizzazione1)
Nella SM, la demielinizzazione è prevalentemente della sostanza bianca con un ruolo centrale dei linfociti T CD8+, mentre nella NMOSD il coinvolgimento dei linfociti T CD4+ è maggiore, formando lesioni necrotiche che interessano sia la sostanza grigia che quella bianca.
Motivo della distribuzione
I canali AQP4 sono abbondantemente distribuiti nel nervo ottico, nell’area postrema e nel midollo spinale, rendendo queste regioni selettivamente bersaglio.
Biomarcatori
GFAP sierico: Riflette il danno astrocitario ed è elevato durante gli attacchi.
Catena leggera del neurofilamento sierico (NfL): Riflette il danno assonale ed è correlata alla gravità degli attacchi1)
Le citochine coinvolte includono IL-6, IL-10, IL-17a, G-CSF, TNF-α e BAFF/APRIL.
7. Ricerche recenti e prospettive future (rapporti in fase di ricerca)
Si stima che il 3-5% delle NMOSD siano paraneoplastiche. I casi associati a teratoma ovarico sono stati studiati in modo particolarmente dettagliato.
Ikeguchi et al. (2021) hanno condotto una revisione di 6 casi di NMOSD AQP4+ associata a teratoma ovarico 2). Tutti i pazienti erano donne, età media di insorgenza 32,7 anni (15-50 anni). Dell’83% (5/6) dei casi presentavano nausea/vomito, l’83% aveva bande oligoclonali positive nel liquido cefalorachidiano e l’83% presentava lesioni del tronco encefalico dorsale. L’analisi patologica ha confermato l’immunoreattività AQP4 e l’infiltrazione linfocitaria nel tessuto nervoso GFAP-positivo all’interno del tumore, suggerendo un meccanismo in cui la presentazione dell’antigene AQP4 nel tumore scatena una reazione autoimmune. Dopo la resezione del tumore, l’AQP4-IgG è diventato negativo nel 60% (3/5) dei pazienti.
Ding et al. (2021) hanno condotto una revisione di 43 casi di NMOSD paraneoplastica 3). L’88,4% erano donne e i tipi di tumore più frequenti erano il cancro al seno e il cancro al polmone. L’importanza dello screening tumorale è sottolineata in particolare nei pazienti con NMOSD di età pari o superiore a 50 anni.
Lo screening per tumori, inclusi i teratomi, è raccomandato anche nei pazienti giovani.
Terapia di immunoassorbimento per i casi refrattari
L’efficacia dell’immunoassorbimento su proteina A (IA) è stata riportata per la NMOSD refrattaria che non risponde a steroidi, plasmaexchange o rituximab.
Fan et al. (2024) hanno eseguito 3 sedute di immunoassorbimento su proteina A in una donna di 35 anni con NMOSD refrattaria associata a sindrome di Sjögren, non responsiva a boli di steroidi e IVIG4). Entro una settimana, i disturbi visivi, la paraplegia e i disturbi propriocettivi sono migliorati significativamente, con una rapida diminuzione di AQP4-IgG, IgA, IgG e IgM. Durante 4 anni di follow-up non sono state osservate recidive o progressioni.
Caratteristiche dei casi con comorbidità di malattie autoimmuni
La realtà della comorbidità di varie malattie autoimmuni con la NMOSD sta diventando sempre più chiara.
Zhu et al. (2025) hanno riportato il caso di una ragazza di 14 anni che ha sviluppato NMOSD all’età di 11 anni 5). AQP4-IgG positiva, nel corso della malattia è stata confermata la comorbidità con la sindrome di Sjögren primaria. La remissione è stata mantenuta con metilprednisolone, IVIG e micofenolato mofetile (MMF) con successivo passaggio a tacrolimus. Sebbene nei casi adulti di NMOSD sia riportata una comorbidità con malattie autoimmuni nel 20-30%, è stato dimostrato che tale comorbidità esiste anche nei bambini 5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, Suzuki M, Shimoji K, Motohashi T, Yamamoto T, Shibata N, Kitagawa K. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045. doi:10.1212/NXI.0000000000001045. PMID: 34285095. PMCID: PMC8293286.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain and behavior. 2021;11(10):e2282. doi:10.1002/brb3.2282. PMID:34520629; PMCID:PMC8553315.
Fan W, Chen X, Xiao P, Wei B, Zhang Y, Huang J, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Frontiers in immunology. 2024;15:1429405. doi:10.3389/fimmu.2024.1429405. PMID:39055718; PMCID:PMC11269126.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.