La neurite ottica è una malattia in cui il nervo ottico si infiamma per qualche causa, con conseguente riduzione della funzione visiva. Dopo aver escluso le neuropatie ottiche infettive e non infettive (tossiche, ereditarie, compressive), la maggior parte dei casi è idiopatica, probabilmente dovuta a un meccanismo autoimmune.
L’età di esordio è tra 15 e 45 anni, più frequente nelle donne, e si manifesta come neuropatia ottica acuta monoculare. Il deficit visivo progredisce da alcuni giorni a due settimane, quindi mostra una tendenza al recupero entro cinque settimane.
L’incidenza annuale in Giappone è di 1,6 per 100.000 adulti. L’età di esordio è tra 15 e 45 anni, con circa il 70% di donne. La prevalenza mondiale è stimata tra 1 e 5 per 100.000 persone.
L’associazione con la SM è molto stretta: la probabilità cumulativa di sviluppare SM entro 15 anni da una neurite ottica idiopatica è del 50%. In assenza di lesioni demielinizzanti alla RM cerebrale iniziale, il tasso di conversione è solo del 25%, ma se è presente almeno una lesione, raggiunge il 78%.
Nei bambini, l’età di picco è 9-10 anni. Più il bambino è piccolo, più frequente è il coinvolgimento bilaterale e grave la perdita visiva, specialmente sotto i 5 anni. Il tasso di conversione alla SM pediatrica è riportato da Adachi et al. intorno al 30% e da Mizota et al. al 16%.
L’incidenza annuale di MOGAD è stimata tra 1,6 e 4,8 per 1 milione di persone. 1)
QSe sviluppo una neurite ottica, in futuro avrò la sclerosi multipla?
A
Circa il 50% delle neuriti ottiche idiopatiche evolve in SM entro 15 anni. Tuttavia, in assenza di lesioni demielinizzanti alla RM cerebrale iniziale, il tasso di conversione è solo del 25%. Dopo la diagnosi, sono importanti la valutazione del rischio tramite RM cerebrale e il follow-up in collaborazione con un neurologo.
Wikimedia Commons. File:Optic_Neuritis.png. License: CC BY-SA.
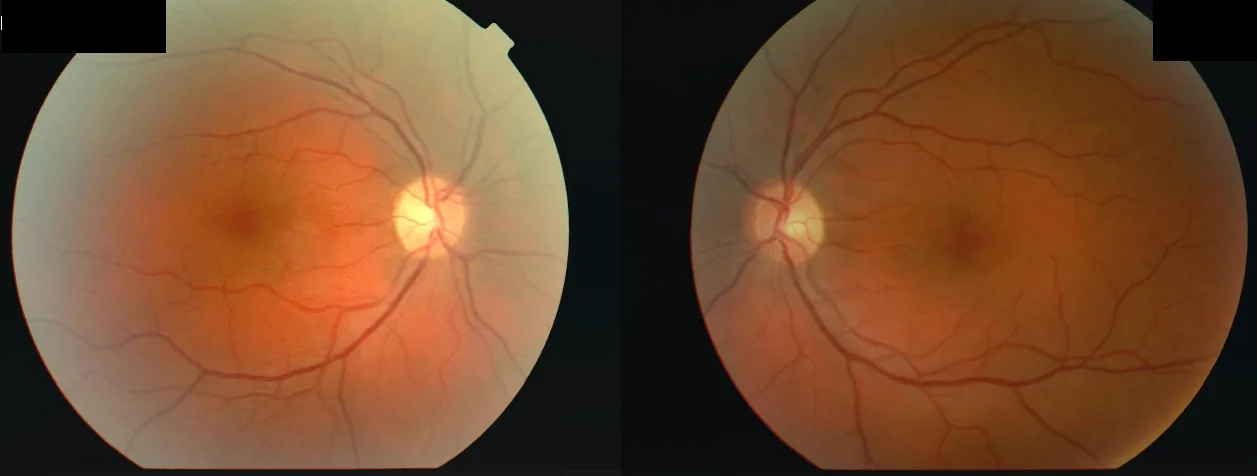
Fotografie del fondo oculare di entrambi gli occhi di una donna di 34 anni che ha presentato una riduzione dell’acuità visiva all’occhio destro (OD 2/60) e poi al sinistro (OS 6/24) dopo una malattia febbrile, con scotoma centrale bilaterale e difetto pupillare afferente relativo (RAPD) all’occhio destro. Ciò corrisponde allo «scotoma centrale» descritto nella sezione «2. Principali sintomi e segni clinici».
L’esordio è acuto, con i seguenti sintomi caratteristici:
Riduzione acuta unilaterale della vista: progressione in pochi giorni fino a 2 settimane. La funzione visiva è peggiore circa una settimana dopo l’esordio.
Dolore oculare o dolore ai movimenti oculari: presente in circa il 50-60% dei casi in Giappone. Può precedere la riduzione della vista di alcuni giorni.
Difetti del campo visivo: spesso scotoma centrale o centro-cecale, ma possibile anche emianopsia orizzontale. Sono comuni anche difetti diffusi.
Anomalie della visione dei colori: riduzione della saturazione del rosso, caratteristica.
Segno di Uhthoff: riduzione transitoria della vista o offuscamento dopo aumento della temperatura corporea (bagno caldo, esercizio fisico). Compare in pochi minuti e scompare entro un’ora.
Fosfeni (phosphenes): percezione di lampi di luce
Fenomeno di Pulfrich: alterazione della percezione della profondità di oggetti in movimento
Difetto pupillare afferente relativo (RAPD): reperto più importante. Positivo in caso di interessamento unilaterale o asimmetrico. Nella neurite ottica è anomalo anche con disfunzione lieve. Da verificare prima della dilatazione pupillare con lente a contatto e lampada a fessura
Grado di deficit visivo: secondo l’ONTT, il 10% ha acuità ≥ 1,0, il 25% tra 0,5 e 1,0, il 29% tra 0,1 e 0,5, il 36% inferiore
Reperti del fondo oculare: nella neurite ottica anteriore si osservano arrossamento e gonfiore della papilla, iperfluorescenza all’angiografia con fluoresceina. Nella neurite ottica retrobulbare inizialmente normale, pallore dopo 4-6 settimane
Oftalmoplegia internucleare: compare nei casi associati a SM per demielinizzazione del fascicolo longitudinale mediale
Guaine vascolari retiniche e uveite periferica: nel 5-10% dei pazienti con SM
Nella neurite ottica pediatrica, si osserva più spesso una papillite tipica con arrossamento e gonfiore della papilla ottica rispetto alla neurite retrobulbare osservata negli adulti. L’infiammazione non si limita alla sola papilla ma si estende per tutta la lunghezza del nervo ottico. È bilaterale nel 50-75% dei casi e la forma di neurite ottica anteriore rappresenta il 50-75% dei casi.
QCos'è il segno di Uhthoff?
A
Il segno di Uhthoff è un fenomeno di deficit visivo transitorio in seguito all’aumento della temperatura corporea. Dopo un bagno o un esercizio fisico, il paziente avverte una riduzione dell’acuità visiva o offuscamento. Compare dopo pochi minuti e scompare entro un’ora. È noto nella neurite ottica associata a SM, ma è stato riportato anche in altre neuropatie ottiche e non è specifico per la SM.
QIn cosa differisce la neurite ottica con anticorpi anti-AQP4 dalla neurite ottica tipica?
A
La neurite ottica con anticorpi anti-AQP4 è una forma refrattaria che rappresenta circa il 10% delle neuriti ottiche idiopatiche. Rispetto alla neurite ottica tipica, l’acuità visiva all’esordio è peggiore, la risposta alla terapia steroidea è scarsa e le recidive sono più frequenti. Differisce anche per la tendenza a diventare bilaterale. La misurazione degli anticorpi sierici anti-AQP4 è essenziale per la diagnosi.
Nei casi idiopatici sospettati di meccanismo autoimmune, cellule infiammatorie come la microglia infiltrano il nervo ottico e causano infiammazione. Infiammazioni ripetute portano a un accumulo di danni alle fibre nervose, con conseguente atrofia ottica.
È stata proposta l’ipotesi che un’infezione virale scateni una reazione autoimmune e in alcuni pazienti vengono rilevati anticorpi virali contro morbillo, varicella, influenza, ecc. nel liquido cerebrospinale. Nei bambini è nota un’associazione con ADEM (encefalomielite acuta disseminata) accompagnata da febbre e cefalea.
Sesso: più frequente nelle donne (rapporto maschi:femmine circa 1:2–1:3)
Età: più comune tra i 20 e i 45 anni
Sintomi prodromici: può essere preceduto da una malattia simil-influenzale
Pazienti con SM: fino al 75% sperimenta almeno un episodio di ON nella vita. All’autopsia, fino al 90% presenta lesioni del nervo ottico.
Vaccinazioni e infezioni sono state segnalate come fattori scatenanti della MOGAD e si ipotizzano meccanismi di rottura della tolleranza immunitaria come l’attivazione di bystander. 1)
Sono stati riportati casi di ON dopo infezione da COVID-19 o vaccinazione. Il tempo mediano dalla vaccinazione all’insorgenza di ON è di 18 giorni e 14 dei 55 casi erano positivi per MOG-IgG. 11)
La neurite ottica è una diagnosi clinica. Viene diagnosticata dalla combinazione di riduzione acuta della vista monolaterale, dolore ai movimenti oculari, RAPD positivo e difetto del campo visivo.
RMN orbitaria : sequenze STIR con soppressione del grasso in coronale e T1 pesate con contrasto. Valutare aumento di volume, iperintensità e enhancement del nervo ottico
RMN encefalica FLAIR : valutare la presenza di lesioni demielinizzanti cerebrali. Essenziale per valutare l’associazione con SM e il rischio futuro di sviluppare SM
La NO tipica e la NO atipica differiscono nei reperti RMN come segue.
Misurazione dello spessore dello strato di fibre nervose retiniche peripapillari (pRNFL). Nella fase acuta si osserva un ispessimento della pRNFL (mediana MOG-ON 164 μm vs MS-ON 103 μm). Nella fase cronica si verifica un assottigliamento della pRNFL e il numero di recidive è correlato alla diminuzione della pRNFL. Al di sotto di una soglia di pRNFL di 50 μm, la deviazione media del campo visivo peggiora significativamente. 1)
In caso di decorso atipico, si misurano i seguenti parametri.
Anticorpi anti-AQP4: coperti dall’assicurazione sanitaria dal 2013. In caso di resistenza agli steroidi o recidive frequenti, è importante una valutazione precoce.
Anticorpi anti-MOG (MOG-IgG): si raccomanda un test sierico con saggio su cellule vive (live cell-based assay). I CBA fissi hanno sensibilità e specificità inferiori.
Secondo i criteri diagnostici internazionali MOGAD 2023, la diagnosi è confermata da CBA positivo + fenotipo clinico centrale + esclusione di diagnosi alternative. In caso di titolo basso, è necessaria almeno una caratteristica clinica/RM di supporto 2).
Validazione di questi criteri: sensibilità 96,5%, specificità 98,9%, accuratezza 98,5% 3).
Valutare la possibilità che la neurite ottica sia il primo sintomo di SM. La diagnosi si basa sulla dimostrazione della disseminazione temporale e spaziale delle lesioni infiammatorie demielinizzanti del sistema nervoso centrale e sull’esclusione di altre malattie. La disseminazione spaziale può essere dimostrata dalla presenza di lesioni iperintense in T2 in almeno due delle quattro regioni: periventricolare, sottocorticale, sottotentoriale e midollo spinale.
È stato riportato un caso diagnosticato come neurite ottica retrobulbare che in realtà era una neuropatia ottica compressiva dovuta a linfoma orbitario. In presenza di caratteristiche atipiche, non somministrare steroidi con leggerezza e procedere a un esame approfondito. 9)
Di solito, la FA (angiografia retinica con fluoresceina), la CFF e il campo visivo dinamico eseguiti negli adulti non sono adatti ai bambini. La diagnosi si basa sui reperti del fundus e sulla RMN cerebrale.
QQuali sono i punti chiave alla RMN per distinguere la neurite ottica tipica da quella atipica?
A
Nella ON tipica, si osserva un potenziamento del contrasto solo in un breve segmento del nervo ottico. Nella ON atipica, invece, si osserva un potenziamento del contrasto esteso del nervo ottico (più della metà della sua lunghezza), estensione posteriore (chiasma ottico, tratto ottico) e potenziamento della guaina del nervo ottico. Questi reperti RMN sono direttamente collegati alla diagnosi differenziale della malattia causale.
La prognosi visiva della neurite ottica idiopatica è buona. In oltre il 90% delle neuriti ottiche, l’acuità visiva migliora con l’osservazione o la somministrazione sistemica di steroidi. Secondo i risultati a lungo termine dell’ONTT, l’acuità visiva a 1 anno dall’esordio è pari o superiore a 0,5 nel 93% dei casi e pari o superiore a 1,0 in oltre il 70%.
Nella neurite ottica idiopatica con acuità visiva corretta relativamente buona, si può optare per l’osservazione con mecobalamina 1.500 μg/die per via orale (non coperto da assicurazione).
ON tipica
Prima scelta: Terapia con bolo di steroidi (metilprednisolone 1.000 mg/die in infusione endovenosa per 3 giorni). Non coperto da assicurazione.
Dopo il bolo: Iniziare prednisolone per via orale a 0,5 mg/kg/die, quindi ridurre gradualmente di 5-10 mg ogni 3-4 giorni.
Se il primo ciclo è inefficace: Eseguire un secondo bolo dopo un intervallo di 4-5 giorni.
Effetto: Riduce il periodo di recupero, ma non vi è differenza significativa nell’acuità visiva finale a 1 anno.
Indicazione attiva : Coinvolgimento bilaterale, grave deficit visivo, unico occhio funzionante, recidiva, placche di demielinizzazione alla RMN, o forte desiderio del paziente di un rapido miglioramento.
Neurite ottica con anticorpi anti-AQP4 positivi
Fase acuta : Terapia con boli di steroidi (prima linea).
Se inefficace : Nuovo bolo dopo 3-4 giorni → se ancora inefficace → considerare plasmaferesi.
Plasmaferesi : Scambio plasmatico semplice > plasmaferesi a doppia filtrazione > immunoassorbimento (in ordine di efficacia). 1 ciclo di 5-6 sedute.
Attenzione : A differenza della neurite ottica idiopatica, la prevenzione delle recidive con steroidi orali è importante.
Neurite ottica con anticorpi anti-MOG positivi (MOGAD)
Fase acuta : La terapia con boli di steroidi è altamente efficace. Recupero completo 50%, parziale 44%. 13)
Tendenza alle recidive : Forte dipendenza da steroidi, 70% di recidive durante la riduzione del prednisolone orale (soprattutto <10 mg/die o entro 2 mesi dalla sospensione). 14)
Terapia di mantenimento : IVIg (≥1 g/kg/4 settimane riduce significativamente le recidive). 15)Rituximab può essere meno efficace rispetto all’AQP4-ON.
Inizio del trattamento : Di solito dopo il secondo attacco (poiché >50% dei casi sono monofasici).
La monoterapia con prednisolone orale a dose standard (1 mg/kg/die) non è raccomandata poiché nello studio ONTT ha mostrato un tasso di recidiva più elevato rispetto al placebo o agli steroidi endovenosi.
Eseguire una terapia steroidea aggressiva. In caso di scarsa risposta al pulse di steroidi, le immunoglobuline endovenose ad alte dosi e la plasmaferesi sono opzioni.
Nei casi associati a SM, dopo il miglioramento visivo, considerare un trattamento per la prevenzione delle recidive. In collaborazione con un neurologo, considerare farmaci modificanti la malattia (interferone beta, glatiramer acetato, fingolimod, natalizumab, ecc.).
QIl trattamento steroideo influisce sul recupero visivo finale?
A
Secondo i risultati dell’ONTT, la terapia pulsata con steroidi accelera la velocità di recupero, ma non ha un effetto significativo sull’acuità visiva finale a un anno. Nella neurite ottica idiopatica, anche senza trattamento, il 93% dei pazienti recupera un’acuità visiva di 0,5 o superiore. Tuttavia, i casi positivi per anticorpi anti-AQP4 sono resistenti agli steroidi e richiedono un trattamento aggressivo precoce, inclusa la plasmaferesi.
QPer quanto tempo deve essere continuata la terapia immunosoppressiva?
A
La CRION è una malattia che recidiva con la sospensione della terapia, pertanto spesso è necessaria un’immunosoppressione a lungo termine anche dopo la remissione dei sintomi. Dopo aver determinato la dose minima efficace di steroidi, si aggiungono farmaci immunosoppressivi non steroidei per progettare un regime terapeutico individualizzato.
Nella forma idiopatica sospetta autoimmune, cellule infiammatorie come la microglia infiltrano il nervo ottico e causano infiammazione.
Distruzione immuno-mediata della guaina mielinica: La guaina mielinica del nervo ottico viene attaccata da un meccanismo autoimmune. La conduzione saltatoria diventa impossibile, causando un disturbo della conduzione dell’impulso assonico.
Degenerazione assonale: Dopo la distruzione della mielina, gli assoni delle cellule gangliari retiniche iniziano a degenerare.
Fagocitosi da parte dei macrofagi: I macrofagi rimuovono i residui di mielina.
Gliosi: Gli astrociti proliferano e formano una cicatrice gliale. Il termine ‘sclerosi’ nella sclerosi multipla deriva da questo processo.
Episodi ripetuti di infiammazione del nervo ottico portano a un danno cumulativo delle fibre nervose, con conseguente atrofia ottica.
Gli anticorpi anti-AQP4 si legano al complemento e attaccano gli astrociti, le cellule gliali del nervo ottico, scatenando la malattia. Gli astrociti del nervo ottico e del chiasma esprimono molto AQP4, rendendoli bersagli vulnerabili.
Meccanismo della neurite ottica positiva per anticorpi anti-MOG
Le MOG-IgG (principalmente sottoclasse IgG1) hanno come bersaglio la MOG sulla superficie della guaina mielinica del SNC. L’attivazione della via del complemento e la fagocitosi cellulare anticorpo-dipendente sono coinvolte nella demielinizzazione. Tuttavia, l’attivazione del complemento è più debole rispetto alle AQP4-IgG. Nella MOGAD predominano i linfociti T CD4+ e i macrofagi, mentre nella SM predominano i linfociti T CD8+. 1)4)
Nonostante la MOG non sia espressa nella retina, nella MOG-ON si verifica un danno alle cellule gangliari retiniche. Come meccanismi sono proposti l’eccitotossicità del glutammato e la fragilità della barriera emato-encefalica a livello della testa del nervo ottico. 1)
È anche in studio il meccanismo mediante il quale l’IL-6 aumenta la permeabilità della barriera emato-encefalica e promuove la differenziazione dei plasmablasti.
Il CRION è stato originariamente proposto come una sindrome che comprende neuriti ottiche corticosensibili e recidivanti. 5) Successivi test anticorpali in coorti CRION hanno mostrato che fino al 22% erano positivi per AQP4-IgG e fino al 25% per MOG-IgG. 6)7)8) Pertanto, il CRION è considerato una diagnosi sindromica che comprende un gruppo eterogeneo di eziologie, inclusa la neurite ottica associata ad anticorpi MOG e la neurite ottica associata ad anticorpi AQP4.
È stata proposta l’ipotesi che un’infezione virale scateni una reazione autoimmune. Sono stati riportati casi di rilevamento di DNA del virus varicella-zoster e del virus herpes simplex nel liquido cerebrospinale di alcuni pazienti con NO. Nella MOGAD si ipotizza una rottura della tolleranza immunitaria (attivazione bystander, mimetismo molecolare) dopo vaccinazione o infezione. 1)
Per la NMOSD positiva per AQP4 sono disponibili agenti biologici con evidenza di livello 1. 4)
Eculizumab: inibitore del C5. Nella fase 3, riduzione del rischio di recidiva del 94%.
Ravulizumab: inibitore del C5 (versione migliorata di eculizumab). Nella fase 3, riduzione del rischio di recidiva del 98,6%.
Inebilizumab: farmaco depletore delle cellule B mirato al CD19. Riduzione del 77% del rischio di recidiva nella NMOSD AQP4-positiva.
Satralizumab: inibitore del recettore dell’IL-6. Riduzione del 74-79% del rischio di recidiva nella NMOSD AQP4-positiva. Somministrazione sottocutanea possibile a domicilio.
Tutti questi hanno evidenza consolidata per la NMOSD AQP4-positiva, ma l’indicazione per la MOGAD non è ancora stabilita.
Sono in corso studi di fase 3 per satralizumab (NCT05271409) e rozanolixizumab (NCT05063162). L’uso off-label di tocilizumab (anticorpo anti-recettore dell’IL-6) ha mostrato un effetto di prevenzione delle recidive fino a 29 mesi. 1)
Sono stati riportati casi di neurite ottica demielinizzante dopo infezione da COVID-19. Jossy et al. (2022) hanno riportato casi di neurite ottica insorta durante la fase di convalescenza, tutti con recupero della vista dopo terapia pulsata con steroidi. 10)
Si stanno accumulando segnalazioni di neurite ottica dopo vaccinazione anti-COVID-19, con un intervallo mediano di 18 giorni dalla vaccinazione all’insorgenza della NO. Su 55 casi, 14 erano MOG-IgG positivi e nessuno AQP4-IgG positivo. 11) Sono stati riportati anche casi di neurite ottica associata ad anticorpi MOG dopo infezione da SARS-CoV-2. 12)
Con la diffusione dei test per gli anticorpi MOG-IgG e AQP4-IgG, alcuni casi precedentemente diagnosticati come CRION vengono riclassificati come MOGAD o NMOSD. In futuro, si prevede che la ridefinizione dei concetti di malattia basata sul profilo anticorpale progredirà.
Jeyakumar N, Lerch M, Dale RC, Ramanathan S. MOG antibody-associated optic neuritis. Eye (London, England). 2024;38(12):2289-2301. doi:10.1038/s41433-024-03108-y. PMID:38783085; PMCID:PMC11306565.
Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. The Lancet. Neurology. 2023;22(3):268-282. doi:10.1016/S1474-4422(22)00431-8. PMID:36706773.
Varley JA, Champsas D, Prossor T, et al. Validation of the 2023 International Diagnostic Criteria for MOGAD in a Selected Cohort of Adults and Children. Neurology. 2024.
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment: A Tale of Two Central Nervous System Autoimmune Inflammatory Disorders. Neurol Clin. 2024;42(1):77-114. doi:10.1016/j.ncl.2023.06.009. PMID:37980124; PMCID:PMC10658081.
Petzold A, Woodhall M, Khaleeli Z, Tobin WO, Pittock SJ, Weinshenker BG, et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein antibodies in immune-mediated optic neuritis at long-term follow-up. Journal of neurology, neurosurgery, and psychiatry. 2019;90(9):1021-1026. doi:10.1136/jnnp-2019-320493. PMID:31118222.
Lee HJ, Kim B, Waters P, Woodhall M, Irani S, Ahn S, et al. Chronic relapsing inflammatory optic neuropathy (CRION): a manifestation of myelin oligodendrocyte glycoprotein antibodies. Journal of neuroinflammation. 2018;15(1):302. doi:10.1186/s12974-018-1335-x. PMID:30382857; PMCID:PMC6208174.
Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT. Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. Journal of neurology, neurosurgery, and psychiatry. 2010;81(1):109-11. doi:10.1136/jnnp.2008.146894. PMID:20019228.
McElhinney K, Rhatigan M, Tsvetanova Z, O’Keane C, Logan P. Beware the Retrobulbar Optic Neuritis Diagnosis. Case reports in ophthalmology. 2022;13(2):453-458. doi:10.1159/000524685. PMID:35950025; PMCID:PMC9247490.
Jossy A, Jacob N, Sarkar S, Gokhale T, Kaliaperumal S, Deb AK. COVID-19-associated optic neuritis - A case series and review of literature. Indian journal of ophthalmology. 2022;70(1):310-316. doi:10.4103/ijo.IJO_2235_21. PMID:34937266; PMCID:PMC8917537.
Bhatti MT, Gilbert AL, Watson G, et al. Shot in the dark. Surv Ophthalmol. 2023;68:821-829.
Francesca Bosello, Damiano Marastoni, Francesca Benedetta Pizzini, Chiara Zaffalon, Andrea Zuliani, Giulia Turri, Sara Mariotto, Erika Bonacci, et al. Atypical myelin oligodendrocyte glycoprotein antibody–associated optic neuritis and acute demyelinating polyneuropathy after SARS-CoV-2 infection: Case report and literature review. Journal of Neuroimmunology. 2023;375:578011. doi:10.1016/j.jneuroim.2022.578011.
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. Journal of neuroinflammation. 2016;13(1):280. doi:10.1186/s12974-016-0718-0. PMID:27793206; PMCID:PMC5086042.
Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. Journal of neurology, neurosurgery, and psychiatry. 2018;89(2):127-137. doi:10.1136/jnnp-2017-316880. PMID:29142145; PMCID:PMC5800335.
Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. MOG antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8-15.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.