La névrite optique est une maladie dans laquelle le nerf optique s’enflamme pour une raison quelconque, entraînant une diminution de la fonction visuelle. Après avoir exclu les neuropathies optiques infectieuses et non infectieuses (toxiques, héréditaires, compressives), la majorité des cas sont idiopathiques, probablement dus à un mécanisme auto-immun.
L’âge de prédilection est de 15 à 45 ans, plus fréquent chez les femmes, et se manifeste par une neuropathie optique aiguë monoculaire. Le déficit visuel progresse sur quelques jours à deux semaines, puis montre une tendance à la récupération dans les cinq semaines.
L’incidence annuelle au Japon est de 1,6 pour 100 000 adultes. L’âge de prédilection est de 15 à 45 ans, avec environ 70% de femmes. La prévalence mondiale est estimée entre 1 et 5 pour 100 000 personnes.
L’association avec la SEP est très étroite : le risque cumulé de développer une SEP dans les 15 ans suivant une névrite optique idiopathique est de 50 %. En l’absence de lésions démyélinisantes à l’IRM cérébrale initiale, ce risque n’est que de 25 %, mais il atteint 78 % si au moins une lésion est présente.
Chez l’enfant, l’âge de prédilection est de 9 à 10 ans. Plus l’enfant est jeune, plus l’atteinte est bilatérale et sévère, surtout avant 5 ans. Le taux de conversion vers la SEP pédiatrique est rapporté à environ 30 % par Adachi et al., et à 16 % par Mizota et al.
L’incidence annuelle du MOGAD est estimée entre 1,6 et 4,8 pour 1 million de personnes. 1)
QSi je développe une névrite optique, vais-je développer une sclérose en plaques à l'avenir ?
A
Environ 50 % des névrites optiques idiopathiques évoluent vers une SEP dans les 15 ans. Cependant, en l’absence de lésions démyélinisantes à l’IRM cérébrale initiale, ce taux n’est que de 25 %. Après le diagnostic, une évaluation du risque par IRM cérébrale et un suivi en collaboration avec un neurologue sont essentiels.
Wikimedia Commons. File:Optic_Neuritis.png. License: CC BY-SA.
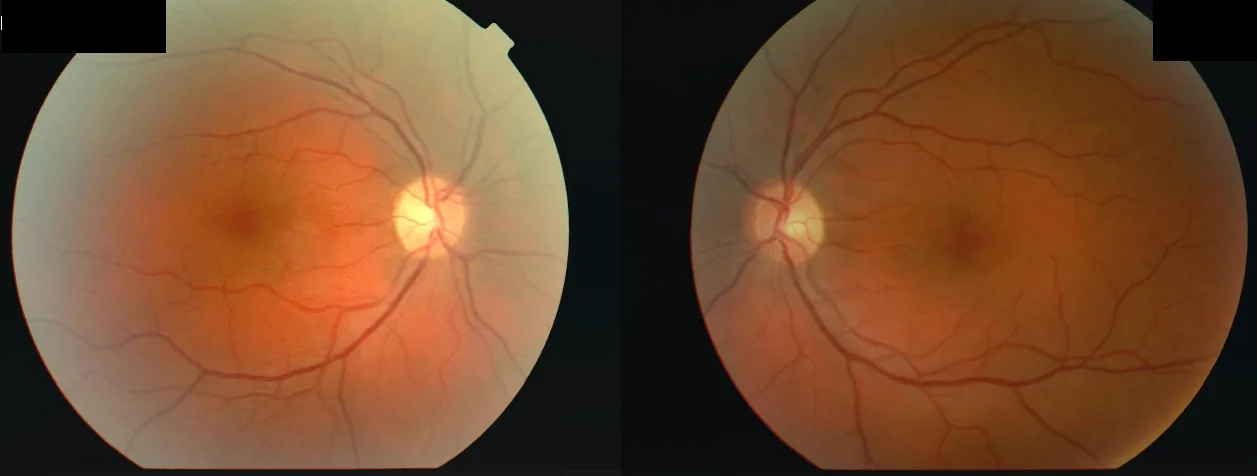
Fond d’œil d’une femme de 34 ans ayant présenté une baisse d’acuité visuelle de l’œil droit (OD 2/60) puis de l’œil gauche (OS 6/24) après une maladie fébrile, avec un scotome central bilatéral et un déficit pupillaire afférent relatif (DPAR) de l’œil droit. Cette image illustre le « scotome central » décrit dans la section « 2. Principaux symptômes et signes cliniques ».
Le mode d’installation est aigu, avec les symptômes caractéristiques suivants :
Baisse d’acuité visuelle aiguë unilatérale : progression sur quelques jours à 2 semaines, avec un maximum de sévérité vers la première semaine.
Douleur oculaire ou douleur aux mouvements oculaires : présente dans environ 50 à 60 % des cas au Japon, pouvant précéder la baisse d’acuité visuelle de quelques jours.
Déficit du champ visuel : souvent un scotome central ou centro-cæcal, mais aussi une hémianopsie horizontale ; les atteintes diffuses sont fréquentes.
Troubles de la vision des couleurs : diminution de la saturation du rouge, caractéristique.
Signe d’Uhthoff : baisse visuelle transitoire ou vision trouble après une élévation de la température corporelle (bain chaud, exercice), apparaissant en quelques minutes et disparaissant en moins d’une heure.
Phosphènes (phosphenes) : perception de flashs lumineux
Phénomène de Pulfrich : modification de la perception de la profondeur des objets en mouvement
Déficit pupillaire afférent relatif (DPAR) : signe le plus important. Positif en cas d’atteinte unilatérale ou asymétrique. Dans la névrite optique, il est anormal même en cas de dysfonctionnement léger. À vérifier avant dilatation avec une lentille de contact et une lampe à fente
Degré de déficit visuel : selon l’ONTT, 10 % ont une acuité ≥ 1,0, 25 % entre 0,5 et 1,0, 29 % entre 0,1 et 0,5, et 36 % en dessous
Fond d’œil : dans la névrite optique antérieure, on observe un œdème et une rougeur de la papille, avec hyperfluorescence à l’angiographie à la fluorescéine. Dans la névrite optique rétrobulbaire, le fond d’œil est normal au début, puis pâleur après 4 à 6 semaines
Ophtalmoplégie internucléaire : apparaît dans les cas associés à la SEP en raison d’une démyélinisation du faisceau longitudinal médian
Gaines vasculaires rétiniennes et uvéite périphérique : chez 5 à 10 % des patients atteints de SEP
Dans la névrite optique pédiatrique, on observe plus souvent une papillite typique avec rougeur et gonflement de la papille optique que la névrite rétrobulbaire observée chez l’adulte. L’inflammation ne se limite pas à la papille mais s’étend sur toute la longueur du nerf optique. L’atteinte est bilatérale dans 50 à 75 % des cas, et la forme de névrite optique antérieure représente 50 à 75 % des cas.
QQu'est-ce que le signe d'Uhthoff ?
A
Le signe d’Uhthoff est un phénomène de trouble visuel transitoire lors d’une augmentation de la température corporelle. Après un bain ou un exercice, le patient ressent une baisse de l’acuité visuelle ou un flou visuel. Il apparaît après quelques minutes et disparaît en moins d’une heure. Il est connu dans la névrite optique associée à la SEP, mais a également été rapporté dans d’autres neuropathies optiques et n’est pas spécifique à la SEP.
QEn quoi la névrite optique avec anticorps anti-AQP4 diffère-t-elle de la névrite optique typique ?
A
La névrite optique avec anticorps anti-AQP4 est une forme réfractaire représentant environ 10 % des névrites optiques idiopathiques. Par rapport à la névrite optique typique, l’acuité visuelle au début est plus faible, la réponse au traitement stéroïdien est moins bonne et les récidives sont plus fréquentes. Elle diffère également par une tendance à évoluer vers une atteinte bilatérale. Le dosage des anticorps sériques anti-AQP4 est indispensable au diagnostic.
Dans les formes idiopathiques suspectées d’être d’origine auto-immune, des cellules inflammatoires comme les microglies infiltrent le nerf optique et provoquent une inflammation. Des inflammations répétées entraînent une accumulation de dommages aux fibres nerveuses, conduisant à une atrophie optique.
L’hypothèse d’une infection virale déclenchant une réaction auto-immune a été proposée, et des anticorps viraux contre la rougeole, la varicelle, la grippe, etc., sont détectés dans le liquide céphalorachidien de certains patients. Chez l’enfant, un lien avec l’ADEM (encéphalomyélite aiguë disséminée) accompagnée de fièvre et de céphalées est connu.
Sexe : plus fréquent chez les femmes (rapport homme/femme d’environ 1:2 à 1:3)
Âge : survient principalement entre 20 et 45 ans
Symptômes prodromiques : peut être précédé d’une maladie pseudo-grippale
Patients atteints de SEP : jusqu’à 75 % présentent au moins un épisode de NO au cours de leur vie. À l’autopsie, jusqu’à 90 % présentent des lésions du nerf optique.
La vaccination et les infections ont été signalées comme déclencheurs du MOGAD, et des mécanismes de rupture de tolérance immunitaire tels que l’activation des bystanders sont suspectés. 1)
Des cas de NO après infection par le COVID-19 ou vaccination ont été rapportés. Le délai médian entre la vaccination et l’apparition de la NO est de 18 jours, et 14 des 55 cas étaient positifs pour les anticorps MOG-IgG. 11)
La névrite optique est un diagnostic clinique. Elle est diagnostiquée par la combinaison d’une baisse de vision aiguë unilatérale, d’une douleur aux mouvements oculaires, d’un RAPD positif et d’un déficit du champ visuel.
IRM orbitaire : séquence STIR avec saturation de graisse en coupe coronale et T1 avec gadolinium. Rechercher un élargissement, un hypersignal et une prise de contraste du nerf optique
IRM cérébrale FLAIR : évaluer la présence de lésions démyélinisantes cérébrales. Essentiel pour évaluer une association avec la SEP et le risque futur de développer une SEP
Les résultats IRM diffèrent entre la NO typique et la NO atypique sur les points suivants.
Mesure de l’épaisseur de la couche des fibres nerveuses rétiniennes péripapillaires (pRNFL). En phase aiguë, on observe un épaississement de la pRNFL (médiane MOG-ON 164 μm vs MS-ON 103 μm). En phase chronique, un amincissement de la pRNFL se produit, et le nombre de récidives est corrélé à la diminution de la pRNFL. En dessous d’un seuil de pRNFL de 50 μm, la déviation moyenne du champ visuel est significativement aggravée. 1)
En cas d’évolution atypique, mesurer les éléments suivants.
Anti-AQP4 anticorps: remboursé par l’assurance maladie en 2013. Une évaluation précoce est importante en cas de résistance aux stéroïdes ou de récidives fréquentes.
Anti-MOG anticorps (MOG-IgG): un test sérique par test cellulaire vivant (live cell-based assay) est recommandé. Les tests CBA fixes ont une sensibilité et une spécificité inférieures.
Selon les critères diagnostiques internationaux MOGAD 2023, le diagnostic est confirmé par CBA positif + phénotype clinique central + exclusion d’autres diagnostics. En cas de faible titre, au moins une caractéristique clinique/IRM de soutien est nécessaire 2).
Validation de ces critères : sensibilité 96,5 %, spécificité 98,9 %, précision 98,5 % 3).
Diagnostic de la SEP selon les critères de McDonald
Évaluer la possibilité que la névrite optique soit le premier symptôme de la SEP. Le diagnostic repose sur la démonstration d’une dissémination temporelle et spatiale des lésions inflammatoires démyélinisantes du système nerveux central, et sur l’exclusion d’autres maladies. La dissémination spatiale peut être démontrée par la présence de lésions en hypersignal T2 dans au moins deux des quatre régions suivantes : périventriculaire, sous-corticale, sous-tentorielle et moelle épinière.
Il a été rapporté qu’un cas diagnostiqué comme névrite optique rétrobulbaire était en réalité une neuropathie optique compressive due à un lymphome orbitaire. En présence de caractéristiques atypiques, il ne faut pas administrer de stéroïdes à la légère et un examen approfondi doit être réalisé. 9)
En général, l’angiographie à la fluorescéine (FA), la fréquence critique de fusion (CFF) et le champ visuel dynamique pratiqués chez l’adulte ne conviennent pas aux enfants. Le diagnostic repose sur les résultats du fond d’œil et de l’IRM cérébrale.
QQuels sont les points clés pour distinguer la névrite optique typique de la névrite optique atypique à l'IRM ?
A
Dans la névrite optique typique, on observe un rehaussement de contraste uniquement sur un court segment du nerf optique. En revanche, dans la névrite optique atypique, on observe un rehaussement de contraste étendu du nerf optique (plus de la moitié de sa longueur), une extension postérieure (chiasma optique, bandelettes optiques) et un rehaussement de la gaine du nerf optique. Ces résultats IRM sont directement liés au diagnostic différentiel de la maladie causale.
Le pronostic visuel de la névrite optique idiopathique est bon. Dans plus de 90 % des cas de névrite optique, l’acuité visuelle s’améliore avec une observation ou une administration systémique de stéroïdes. Selon les résultats à long terme de l’ONTT, l’acuité visuelle un an après le début est de 0,5 ou plus chez 93 % des patients et de 1,0 ou plus chez plus de 70 %.
Dans la névrite optique idiopathique avec une acuité visuelle corrigée relativement bonne, on peut envisager une observation sous méprobamate à 1 500 μg/jour par voie orale (hors AMM).
Névrite optique typique
Premier choix: Traitement par bolus de stéroïdes (méthylprednisolone 1 000 mg/jour en perfusion intraveineuse pendant 3 jours). Hors AMM.
Après le bolus: Commencer la prednisolone par voie orale à 0,5 mg/kg/jour, puis réduire progressivement de 5 à 10 mg tous les 3 à 4 jours.
En cas d’échec de la première cure: Administrer une deuxième cure de bolus après un intervalle de 4 à 5 jours.
Effet: Réduit la durée de récupération, mais il n’y a pas de différence significative dans l’acuité visuelle finale à un an.
Indication active : Atteinte bilatérale, déficience visuelle sévère, œil fonctionnel unique, récidive, lésions de démyélinisation à l’IRM, ou patient souhaitant une amélioration rapide.
Névrite optique avec anticorps anti-AQP4 positif
Phase aiguë : Thérapie par bolus de stéroïdes (première intention).
En cas d’inefficacité : Nouveau bolus après 3-4 jours → si toujours inefficace → envisager une plasmaphérèse.
Plasmaphérèse : Échange plasmatique simple > plasmaphérèse à double filtration > immunoadsorption (par ordre d’efficacité). 1 cure de 5-6 séances.
Attention : Contrairement à la névrite optique idiopathique, la prévention des récidives par corticothérapie orale est importante.
Névrite optique avec anticorps anti-MOG positif (MOGAD)
Phase aiguë : La thérapie par bolus de stéroïdes est très efficace. Récupération complète 50 %, partielle 44 %. 13)
Tendance aux récidives : Forte dépendance aux stéroïdes, 70 % de récidives lors de la réduction de la prednisolone orale (surtout <10 mg/jour ou dans les 2 mois suivant l’arrêt). 14)
Traitement d’entretien : IVIg (≥1 g/kg/4 semaines réduit significativement les récidives). 15) Le rituximab peut être moins efficace que dans l’AQP4-ON.
Début du traitement : Généralement après la deuxième attaque (car >50 % des cas sont monophasiques).
La monothérapie par prednisolone orale à dose standard (1 mg/kg/jour) n’est pas recommandée car elle a montré un taux de récidive plus élevé que le placebo ou les corticostéroïdes intraveineux dans l’étude ONTT.
Effectuer un traitement stéroïdien agressif. En cas de mauvaise réponse au pulse de stéroïdes, les immunoglobulines intraveineuses à haute dose et la plasmaphérèse sont des options.
Dans les cas de SEP associée, envisager un traitement préventif des rechutes après amélioration visuelle. En collaboration avec un neurologue, envisager des traitements de fond (interféron bêta, acétate de glatiramère, fingolimod, natalizumab, etc.).
QLe traitement par stéroïdes affecte-t-il la récupération visuelle finale ?
A
Selon les résultats de l’ONTT, la corticothérapie pulsée accélère la récupération, mais n’a pas d’effet significatif sur l’acuité visuelle finale à un an. Dans la névrite optique idiopathique, même sans traitement, 93 % des patients récupèrent une acuité visuelle d’au moins 0,5. Cependant, les cas positifs aux anticorps anti-AQP4 sont résistants aux stéroïdes et nécessitent un traitement agressif précoce incluant les échanges plasmatiques.
QCombien de temps le traitement immunosuppresseur doit-il être poursuivi ?
A
La CRION étant une maladie qui récidive à l’arrêt du traitement, une immunosuppression à long terme est souvent nécessaire même après la rémission des symptômes. Après avoir déterminé la dose minimale efficace de stéroïdes, on associe des immunosuppresseurs non stéroïdiens pour concevoir un schéma thérapeutique individualisé.
Dans la forme idiopathique suspectée d’être auto-immune, des cellules inflammatoires comme les microglies infiltrent le nerf optique et provoquent une inflammation.
Destruction immunitaire de la gaine de myéline : La gaine de myéline du nerf optique est attaquée par un mécanisme auto-immun. La conduction saltatoire devient impossible, entraînant un trouble de la conduction de l’influx axonique.
Dégénérescence axonale : Après la destruction de la myéline, les axones des cellules ganglionnaires de la rétine commencent à dégénérer.
Phagocytose par les macrophages : Les macrophages éliminent les débris de myéline résiduels.
Gliose : Les astrocytes prolifèrent et forment une cicatrice gliale. Le terme « sclérose » dans la sclérose en plaques provient de ce processus.
Des épisodes répétés d’inflammation du nerf optique entraînent des dommages cumulatifs aux fibres nerveuses, conduisant à une atrophie optique.
Les anticorps anti-AQP4 se lient au complément et attaquent les astrocytes, cellules gliales du nerf optique, déclenchant la maladie. Les astrocytes du nerf optique et du chiasma expriment fortement l’AQP4, ce qui en fait des cibles privilégiées.
Mécanisme de la névrite optique positive aux anticorps anti-MOG
Les MOG-IgG (principalement de sous-classe IgG1) ciblent la MOG à la surface de la gaine de myéline du SNC. L’activation de la voie du complément et la phagocytose cellulaire dépendante des anticorps sont impliquées dans la démyélinisation. Cependant, l’activation du complément est plus faible par rapport aux AQP4-IgG. Dans la MOGAD, les lymphocytes T CD4+ et les macrophages prédominent, tandis que dans la SEP, les lymphocytes T CD8+ sont prédominants. 1)4)
Bien que la MOG ne soit pas exprimée dans la rétine, la MOG-ON entraîne des lésions des cellules ganglionnaires rétiniennes. L’excitotoxicité au glutamate et la fragilité de la barrière hémato-encéphalique au niveau de la tête du nerf optique sont proposées comme mécanismes. 1)
Le rôle de l’IL-6 dans l’augmentation de la perméabilité de la barrière hémato-encéphalique et la promotion de la différenciation des plasmablastes est également étudié.
Le CRION a été initialement proposé comme un syndrome incluant les névrites optiques corticosensibles et récurrentes. 5) Des tests d’anticorps ultérieurs dans des cohortes CRION ont montré que jusqu’à 22 % étaient positifs pour les AQP4-IgG et jusqu’à 25 % pour les MOG-IgG. 6)7)8) Ainsi, le CRION est considéré comme un diagnostic syndromique englobant un groupe hétérogène d’étiologies, y compris la névrite optique associée aux anticorps MOG et la névrite optique associée aux anticorps AQP4.
L’hypothèse selon laquelle une infection virale déclenche une réaction auto-immune a été proposée. Des rapports ont détecté de l’ADN du virus varicelle-zona et du virus herpes simplex dans le liquide céphalorachidien de certains patients atteints de NO. Dans la MOGAD, on suppose une rupture de la tolérance immunitaire (activation bystander, mimétisme moléculaire) suite à une vaccination ou une infection. 1)
Des agents biologiques avec un niveau de preuve 1 sont apparus pour la NMOSD positive aux AQP4. 4)
Éculizumab : inhibiteur du C5. Réduction du risque de rechute de 94 % dans l’essai de phase 3.
Ravulizumab : inhibiteur du C5 (version améliorée de l’éculizumab). Réduction du risque de rechute de 98,6 % dans l’essai de phase 3.
Inebilizumab: médicament de déplétion des cellules B ciblant CD19. Réduction de 77% du risque de rechute dans la NMOSD AQP4-positive.
Satralizumab: inhibition du récepteur de l’IL-6. Réduction de 74 à 79% du risque de rechute dans la NMOSD AQP4-positive. Administration sous-cutanée possible à domicile.
Ces traitements ont tous des preuves établies pour la NMOSD AQP4-positive, mais leur indication pour la MOGAD n’est pas encore établie.
Les essais de phase 3 du satralizumab (NCT05271409) et du rozanolixizumab (NCT05063162) sont en cours. L’utilisation hors AMM du tocilizumab (anticorps anti-récepteur de l’IL-6) a montré un effet de prévention des rechutes jusqu’à 29 mois. 1)
Des cas de névrite optique démyélinisante après une infection par la COVID-19 ont été rapportés. Jossy et al. (2022) ont rapporté des cas de névrite optique survenue pendant la phase de convalescence, tous ayant récupéré leur acuité visuelle après une corticothérapie pulsée. 10)
Des cas de névrite optique après vaccination contre la COVID-19 sont accumulés, avec un délai médian de 18 jours entre la vaccination et l’apparition de la névrite optique. Sur 55 cas, 14 étaient positifs pour les IgG MOG, et aucun pour les IgG AQP4. 11) Des cas de névrite optique associée aux anticorps MOG après infection par le SARS-CoV-2 ont également été rapportés. 12)
Avec la généralisation des tests d’anticorps MOG-IgG et AQP4-IgG, une partie des cas précédemment diagnostiqués comme CRION sont reclassés en MOGAD ou NMOSD. À l’avenir, une redéfinition des concepts de maladie basée sur le profil d’anticorps devrait progresser.
Jeyakumar N, Lerch M, Dale RC, Ramanathan S. MOG antibody-associated optic neuritis. Eye (London, England). 2024;38(12):2289-2301. doi:10.1038/s41433-024-03108-y. PMID:38783085; PMCID:PMC11306565.
Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. The Lancet. Neurology. 2023;22(3):268-282. doi:10.1016/S1474-4422(22)00431-8. PMID:36706773.
Varley JA, Champsas D, Prossor T, et al. Validation of the 2023 International Diagnostic Criteria for MOGAD in a Selected Cohort of Adults and Children. Neurology. 2024.
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment: A Tale of Two Central Nervous System Autoimmune Inflammatory Disorders. Neurol Clin. 2024;42(1):77-114. doi:10.1016/j.ncl.2023.06.009. PMID:37980124; PMCID:PMC10658081.
Petzold A, Woodhall M, Khaleeli Z, Tobin WO, Pittock SJ, Weinshenker BG, et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein antibodies in immune-mediated optic neuritis at long-term follow-up. Journal of neurology, neurosurgery, and psychiatry. 2019;90(9):1021-1026. doi:10.1136/jnnp-2019-320493. PMID:31118222.
Lee HJ, Kim B, Waters P, Woodhall M, Irani S, Ahn S, et al. Chronic relapsing inflammatory optic neuropathy (CRION): a manifestation of myelin oligodendrocyte glycoprotein antibodies. Journal of neuroinflammation. 2018;15(1):302. doi:10.1186/s12974-018-1335-x. PMID:30382857; PMCID:PMC6208174.
Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT. Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. Journal of neurology, neurosurgery, and psychiatry. 2010;81(1):109-11. doi:10.1136/jnnp.2008.146894. PMID:20019228.
McElhinney K, Rhatigan M, Tsvetanova Z, O’Keane C, Logan P. Beware the Retrobulbar Optic Neuritis Diagnosis. Case reports in ophthalmology. 2022;13(2):453-458. doi:10.1159/000524685. PMID:35950025; PMCID:PMC9247490.
Jossy A, Jacob N, Sarkar S, Gokhale T, Kaliaperumal S, Deb AK. COVID-19-associated optic neuritis - A case series and review of literature. Indian journal of ophthalmology. 2022;70(1):310-316. doi:10.4103/ijo.IJO_2235_21. PMID:34937266; PMCID:PMC8917537.
Bhatti MT, Gilbert AL, Watson G, et al. Shot in the dark. Surv Ophthalmol. 2023;68:821-829.
Francesca Bosello, Damiano Marastoni, Francesca Benedetta Pizzini, Chiara Zaffalon, Andrea Zuliani, Giulia Turri, Sara Mariotto, Erika Bonacci, et al. Atypical myelin oligodendrocyte glycoprotein antibody–associated optic neuritis and acute demyelinating polyneuropathy after SARS-CoV-2 infection: Case report and literature review. Journal of Neuroimmunology. 2023;375:578011. doi:10.1016/j.jneuroim.2022.578011.
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. Journal of neuroinflammation. 2016;13(1):280. doi:10.1186/s12974-016-0718-0. PMID:27793206; PMCID:PMC5086042.
Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. Journal of neurology, neurosurgery, and psychiatry. 2018;89(2):127-137. doi:10.1136/jnnp-2017-316880. PMID:29142145; PMCID:PMC5800335.
Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. MOG antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8-15.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.