A neurite óptica é uma doença na qual o nervo óptico inflama por alguma causa, resultando em diminuição da função visual. Após excluir neuropatias ópticas infecciosas e não infecciosas (tóxicas, hereditárias, compressivas), a maioria dos casos é idiopática, supostamente por mecanismo autoimune.
A idade de início comum é 15–45 anos, mais frequente em mulheres, e manifesta-se como neuropatia óptica aguda unilateral. O déficit visual progride por alguns dias a 2 semanas, depois mostra tendência à recuperação dentro de 5 semanas.
A incidência anual no Japão é de 1,6 por 100.000 adultos. A idade de início mais comum é entre 15 e 45 anos, com mulheres representando cerca de 70%. A prevalência global é estimada em cerca de 1 a 5 por 100.000 pessoas.
A associação com EM é muito profunda, a probabilidade cumulativa de transição para EM em 15 anos após o início da neurite óptica idiopática é de 50%. Se não houver lesões desmielinizantes na ressonância magnética cerebral no primeiro episódio, a taxa de transição permanece em apenas 25%, mas se houver uma ou mais lesões, atinge 78%.
Em crianças, a idade de início mais comum é 9-10 anos, e quanto mais jovem a criança, mais frequentemente ocorre envolvimento bilateral e perda visual grave. Isso é particularmente notável em crianças menores de 5 anos. A taxa de transição para EM em crianças foi relatada como cerca de 30% por Adachi e 16% por Mizota.
A incidência anual de MOGAD é estimada em 1,6 a 4,8 por milhão de pessoas. 1)
QA neurite óptica leva à esclerose múltipla no futuro?
A
Acredita-se que cerca de 50% das neurites ópticas idiopáticas evoluam para EM em 15 anos. No entanto, se não houver lesões desmielinizantes na ressonância magnética cerebral no primeiro episódio, a taxa de transição é de apenas 25%. Após o início, é importante realizar avaliação de risco com ressonância magnética cerebral e acompanhamento coordenado com neurologista.
Wikimedia Commons. File:Optic_Neuritis.png. License: CC BY-SA.
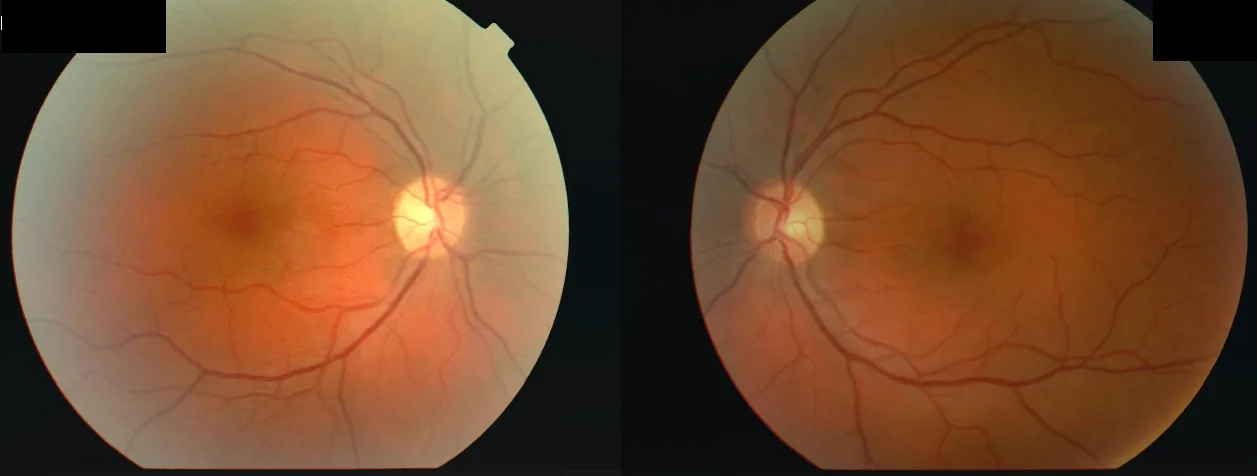
Imagem de fundo de olho de ambos os olhos de uma mulher de 34 anos que apresentou diminuição da acuidade visual no olho direito (OD 2/60) e depois no olho esquerdo (OS 6/24) após doença febril, com escotoma central bilateral e defeito pupilar aferente relativo (RAPD) no olho direito. Isso corresponde ao “escotoma central” discutido na seção “2. Principais sintomas e achados clínicos”.
Apresenta padrão de início agudo, com os seguintes sintomas característicos:
Diminuição aguda da acuidade visual unilateral: Progride ao longo de alguns dias a cerca de 2 semanas. A função visual piora mais cerca de 1 semana após o início
Dor ocular e dor à movimentação ocular: Ocorre em cerca de 50-60% no Japão. Pode aparecer alguns dias antes da diminuição da visão
Distúrbio do campo visual: Escotoma central e escotoma do ponto cego central são comuns, mas também há casos de hemianopsia horizontal. Distúrbio difuso também é comum
Anormalidade da visão de cores: Diminuição da saturação do vermelho é característica
Sinal de Uhthoff: Diminuição temporária da visão ou visão turva com aumento da temperatura corporal (após banho ou exercício). Aparece em minutos e desaparece em uma hora
Fosfenas (phosphenes): Percepção de flashes de luz
Fenômeno de Pulfrich: Alteração na percepção de profundidade de objetos em movimento
Defeito pupilar aferente relativo (RAPD): Achado mais importante. Positivo em casos unilaterais ou assimétricos. Anormal mesmo com disfunção leve na neurite óptica. Verificar com lente de aproximação e lâmpada de fenda antes da dilatação pupilar
Grau de deficiência visual: Segundo o ONTT, 10% têm acuidade de 1,0 ou mais, 25% entre 0,5 e 1,0, 29% entre 0,1 e 0,5 e 36% abaixo disso
Achados de fundo de olho: Na neurite óptica anterior, edema e hiperemia do disco óptico com hiperfluorescência na angiografia fluoresceínica. Na neurite óptica retrobulbar, normal no início, depois palidez após 4-6 semanas
Na neurite óptica infantil, frequentemente se apresenta como papilite típica com vermelhidão e inchaço do disco óptico, em vez da neurite óptica retrobulbar observada em adultos. A inflamação não se limita ao inchaço da papila, mas se estende por todo o comprimento do nervo óptico. O envolvimento bilateral ocorre em 50-75%, e o tipo de neurite óptica anterior representa 50-75%.
QQuais são os sintomas do fenômeno de Uhthoff?
A
O fenômeno de Uhthoff é uma condição em que ocorre deficiência visual transitória com o aumento da temperatura corporal. O paciente percebe diminuição da acuidade visual ou visão turva após banho quente ou exercício. Os sintomas aparecem em minutos e desaparecem dentro de uma hora. É conhecido na neurite óptica associada à EM, mas também é relatado em outras neuropatias ópticas, não sendo específico da EM.
QComo a neurite óptica com anticorpo anti-AQP4 positivo difere da neurite óptica comum?
A
A neurite óptica com anticorpo anti-AQP4 positivo é um tipo refratário que representa cerca de 10% das neurites ópticas idiopáticas. Comparada à neurite óptica comum, a acuidade visual no início é pior, a resposta ao tratamento com esteroides é fraca e as recidivas são mais frequentes. Também difere na tendência a se tornar bilateral. O diagnóstico requer a medição do anticorpo anti-AQP4 no soro.
Nos casos idiopáticos suspeitos de mecanismo autoimune, células relacionadas à inflamação, como micróglia, infiltram o nervo óptico e causam inflamação. A inflamação repetida leva ao acúmulo de danos nas fibras nervosas, resultando em atrofia do nervo óptico.
Há a hipótese de que a infecção viral possa desencadear uma reação autoimune, e anticorpos contra vírus como sarampo, varicela, influenza, etc., são detectados no líquido cefalorraquidiano de alguns pacientes. Em crianças, é conhecida a associação com ADEM (encefalomielite disseminada aguda) acompanhada de febre e dor de cabeça.
Sexo: Mais comum em mulheres (proporção homem:mulher de aproximadamente 1:2 a 1:3)
Idade: Ocorre com mais frequência entre 20 e 45 anos
Sintomas Prodrômicos: Pode ser precedido por uma doença semelhante à gripe
Pacientes com EM: Até 75% experimentam pelo menos um episódio de ON ao longo da vida. Na autópsia, até 90% apresentam lesões do nervo óptico
O MOGAD foi relatado como desencadeado por vacinação ou infecção, e mecanismos como ativação bystander são suspeitos de causar ruptura da tolerância imunológica. 1)
Foram relatados casos de ON após infecção por COVID-19 ou vacinação. O tempo mediano da vacinação ao início da ON foi de 18 dias, e 14 dos 55 casos foram positivos para MOG-IgG. 11)
A neurite óptica é um diagnóstico clínico. É diagnosticada pela combinação de diminuição aguda da visão unilateral, dor ao movimento ocular, RAPD positivo e defeito de campo visual.
Ressonância magnética orbitária: Corte coronal STIR com supressão de gordura, T1 com contraste. Verificar aumento do nervo óptico, hiperintensidade e efeito de contraste.
Ressonância magnética craniana FLAIR: Avaliar presença de lesões desmielinizantes cerebrais. Essencial para avaliar comorbidade com EM e risco futuro de desenvolver EM.
Os achados de RM na ON típica e atípica diferem nos seguintes pontos.
Característica
ON típica (relacionada à EM)
ON atípica
Padrão de contraste
Realce de contraste em segmento curto
Realce de contraste longo e extenso (≥1/2)
Local da lesão
Anterior (nervo óptico retrobulbar)
Estende-se posteriormente (quiasma e trato óptico)
Bainha do nervo óptico
Sem realce por contraste
Realce por contraste da bainha do nervo óptico e gordura orbitária
Comparação das características de imagem da NMOSD-ON, MOG-ON e MS-ON:
Mede a espessura da camada de fibras nervosas da retina peripapilar (pRNFL). Na fase aguda, há espessamento da pRNFL (mediana MOG-ON 164 μm vs MS-ON 103 μm). Na fase crônica, ocorre afinamento da pRNFL, e o número de recidivas correlaciona-se com a redução da pRNFL. Abaixo do limiar de 50 μm de pRNFL, o desvio médio do campo visual piora significativamente.1)
Anticorpo anti-AQP4: Coberto pelo seguro desde 2013. A avaliação precoce é importante em casos de resistência a esteroides ou recidivas frequentes
Anticorpo anti-MOG (MOG-IgG): Recomenda-se teste sérico por ensaio baseado em células vivas (live cell-based assay). O CBA fixo tem sensibilidade e especificidade inferiores
De acordo com os critérios diagnósticos internacionais de MOGAD de 2023, o diagnóstico é confirmado por CBA positivo + fenótipo clínico central + exclusão de diagnósticos alternativos. Em caso de título baixo, é necessária pelo menos uma característica clínica/de RM de suporte2)
Avaliar a possibilidade de neurite óptica ser o sintoma inicial de EM. O diagnóstico é feito demonstrando disseminação temporal e espacial de lesões inflamatórias desmielinizantes no sistema nervoso central e excluindo outras doenças. A disseminação espacial pode ser demonstrada se houver lesões hiperintensas em T2 em duas ou mais das quatro áreas: periventricular, subcortical, infratentorial e medula espinhal.
Acompanhado de febre, cefaleia, alteração da consciência
Há relatos de casos diagnosticados como neurite óptica retrobulbar que na verdade eram neuropatia óptica compressiva devido a linfoma orbitário; portanto, se houver características atípicas, não se deve administrar esteroides levianamente e sim realizar investigação detalhada. 9)
Geralmente, FA (angiografia fluoresceínica), CFF e perimetria dinâmica realizados em adultos não são adequados para crianças; o diagnóstico é feito com base nos achados de fundo de olho e ressonância magnética de crânio.
QQuais são os pontos para diferenciar neurite óptica típica e atípica na RM?
A
Na ON típica, observa-se realce de contraste apenas em um segmento curto do nervo óptico. Já na ON atípica, apresentam-se realce de contraste longo do nervo óptico (mais da metade do comprimento), extensão posterior (quiasma óptico e trato óptico) e realce de contraste da bainha do nervo óptico. Esses achados de RM estão diretamente ligados ao diagnóstico diferencial das doenças causadoras.
O prognóstico da função visual na neurite óptica idiopática é bom. Mais de 90% das neurites ópticas melhoram com observação ou administração sistêmica de esteroides. De acordo com os resultados de longo prazo do ONTT, 93% dos casos alcançam acuidade visual de 0,5 ou mais em 1 ano após o início, e mais de 70% alcançam 1,0 ou mais.
Na neurite óptica idiopática com acuidade visual corrigida relativamente boa, pode-se acompanhar com administração oral de mecobalamina 1500 μg/dia (fora da cobertura do seguro).
ON Típica
Primeira escolha: Terapia com pulsoterapia de esteroide (metilprednisolona 1000 mg/dia IV por 3 dias). Fora da cobertura do seguro.
Após o pulso: Iniciar prednisolona oral 0,5 mg/kg/dia, reduzindo gradualmente 5-10 mg a cada 3-4 dias.
Se não houver resposta ao primeiro pulso: Administrar segundo pulso após 4-5 dias.
Efeito: Encurta o período de recuperação, mas não há diferença significativa na acuidade visual final após 1 ano.
Indicações ativas: Acometimento bilateral, comprometimento visual grave, único olho funcional, casos recorrentes, presença de placas de desmielinização na RM, forte desejo do paciente por melhora precoce.
NO com anticorpo anti-AQP4 positivo
Fase aguda: Terapia com pulsoterapia de corticosteroide (primeira escolha).
Se ineficaz: Repetir pulsoterapia após 3-4 dias → se ainda ineficaz → considerar plasmaférese.
Plasmaférese: Plasmaférese simples > plasmaférese por dupla filtração > imunoadsorção (em ordem de eficácia). Um ciclo de 5-6 sessões.
Terapia de manutenção: Prednisolona 5-10 mg/dia + Azatioprina 50-100 mg/dia.
Nota: Diferente da NO idiopática, a prevenção de recidivas com corticosteroide oral é importante.
NO com anticorpo anti-MOG positivo (MOGAD)
Fase aguda: Pulsoterapia com corticosteroide é altamente eficaz. Recuperação completa 50%, recuperação parcial 44%. 13)
Tendência a recidivas: Dependência de corticosteroide é forte, 70% recidivam durante a redução da dose de prednisolona oral (especialmente <10 mg/dia ou dentro de 2 meses após a suspensão). 14)
Terapia de manutenção: IVIg (≥1 g/kg/4 semanas reduz significativamente as recidivas). 15)Rituximabe pode ser menos eficaz que em AQP4-NO.
Início do tratamento: Geralmente após o segundo ataque (pois >50% são monofásicos).
A monoterapia com prednisolona oral na dose padrão (1 mg/kg/dia) não é recomendada, pois no ONTT mostrou maior taxa de recorrência em comparação com placebo ou corticosteroides intravenosos.
A terapia com corticosteroides é realizada agressivamente. Se a resposta ao pulso de corticosteroide for fraca, imunoglobulina intravenosa em alta dose ou plasmaférese são opções.
Em casos com EM, após melhora visual, considere terapia para prevenção de recorrência. Em coordenação com neurologista, considere medicamentos modificadores da doença (interferon beta, acetato de glatirâmer, fingolimode, natalizumabe, etc.).
QO tratamento com esteroides afeta a recuperação visual final?
A
Os resultados do ONTT mostraram que a pulsoterapia com esteroides acelera a velocidade de recuperação, mas não houve diferença significativa na acuidade visual final um ano após o início. Na neurite óptica idiopática, 93% recuperam para acuidade visual de 0,5 ou melhor sem tratamento. No entanto, casos positivos para anti-AQP4 são resistentes a esteroides, necessitando de tratamento agressivo precoce incluindo plasmaférese.
QPor quanto tempo a terapia imunossupressora deve ser continuada?
A
Como a CRION é uma doença que recidiva com a interrupção do tratamento, muitas vezes é necessária a continuação da terapia imunossupressora a longo prazo mesmo após a remissão dos sintomas. A dose mínima eficaz de esteroides é determinada, então medicamentos imunossupressores não esteroides são adicionados, e o regime de tratamento é projetado individualmente.
Em casos idiopáticos suspeitos de mecanismo autoimune, células relacionadas à inflamação como micróglia infiltram o nervo óptico e causam inflamação.
Destruição da mielina mediada por imunidade: A bainha de mielina do nervo óptico é atacada por mecanismo autoimune. A condução saltatória torna-se impossível, causando distúrbio de condução do impulso axonal.
Degeneração axonal: Após a destruição da mielina, os axônios das células ganglionares da retina começam a degenerar.
Fagocitose por macrófagos: Macrófagos removem os restos de mielina.
Gliose: Astrócitos proliferam e formam cicatriz glial. O termo “esclerose” na esclerose múltipla deriva disso.
Ao experimentar inflamação repetida do nervo óptico, o dano acumulativo às fibras nervosas leva à atrofia do nervo óptico.
O anticorpo anti-AQP4 se liga ao complemento e ataca os astrócitos (células gliais) dentro do nervo óptico, causando o início da doença. Os astrócitos no nervo óptico e quiasma óptico expressam AQP4 em abundância, tornando-se alvos fáceis.
Mecanismo da Neurite Óptica Positiva para Anticorpo Anti-MOG
MOG-IgG (principalmente subclasse IgG1) tem como alvo MOG na superfície da bainha de mielina do SNC. A ativação do complemento e a fagocitose celular dependente de anticorpos estão envolvidas na desmielinização. No entanto, a ativação do complemento é mais fraca em comparação com AQP4-IgG. Na MOGAD, predominam células T CD4-positivas e macrófagos, enquanto na EM, predominam células T CD8-positivas. 1)4)
Embora MOG não seja expresso na retina, a ON associada a MOG causa danos às células ganglionares da retina. Toxicidade por glutamato e fragilidade da barreira hematoencefálica na cabeça do nervo óptico foram propostos como mecanismos. 1)
A IL-6 aumenta a permeabilidade da barreira hematoencefálica e promove a diferenciação de plasmoblastos, um mecanismo também em destaque.
CRION foi originalmente proposto como uma síndrome abrangendo neurite óptica responsiva a esteroides e com tendência a recaídas. 5) Testes de anticorpos subsequentes em coortes de CRION mostraram que até 22% eram positivos para AQP4-IgG e até 25% eram positivos para MOG-IgG. 6)7)8) Portanto, CRION é considerado um diagnóstico sindrômico que engloba um grupo heterogêneo de etiologias, incluindo neurite óptica associada a anticorpos MOG e neurite óptica associada a anticorpos AQP4.
A hipótese de que a infecção viral pode desencadear uma resposta autoimune foi proposta. Há relatos de detecção de DNA do vírus varicela-zoster e do vírus herpes simples no líquido cefalorraquidiano de alguns pacientes com ON. Na MOGAD, vacinação ou infecção são consideradas causas de quebra de tolerância imunológica (ativação bystander ou mimetismo molecular). 1)
Agentes biológicos com evidência de Nível 1 para NMOSD positivo para AQP4 surgiram. 4)
Eculizumabe: Inibidor de C5. Fase 3 mostrou redução de 94% no risco de recaída
Ravulizumabe: Inibidor de C5 (versão melhorada do eculizumabe). Fase 3 mostrou redução de 98,6% no risco de recaída
Inebilizumabe: Medicamento depletor de células B alvo CD19. Reduz o risco de recaída em 77% na NMOSD AQP4-positivo.
Satralizumabe: Inibidor do receptor de IL-6. Reduz o risco de recaída em 74-79% na NMOSD AQP4-positivo. Pode ser administrado por via subcutânea em casa.
Todos esses medicamentos têm evidências estabelecidas para NMOSD AQP4-positivo, mas a indicação para MOGAD ainda não foi estabelecida.
Ensaios clínicos de fase 3 (RCT) estão em andamento para satralizumabe (NCT05271409) e rozanolixizumabe (NCT05063162). O uso off-label de tocilizumabe (anticorpo do receptor de IL-6) relatou efeito preventivo de recaída por até 29 meses. 1)
Casos de neurite óptica desmielinizante após infecção por COVID-19 foram relatados. Jossy et al. (2022) relataram casos que desenvolveram neurite óptica durante a convalescença, e todos recuperaram a visão com pulsoterapia com esteroides. 10)
Relatos de neurite óptica após vacinação contra COVID-19 estão se acumulando, com mediana de 18 dias entre a vacinação e o início da neurite óptica. Das 55 casos, 14 foram positivos para MOG-IgG, e nenhum foi positivo para AQP4-IgG. 11) Casos de neurite óptica associada a MOG após infecção por SARS-CoV-2 também foram relatados. 12)
Com a disseminação dos testes de anticorpos MOG-IgG e AQP4-IgG, alguns casos anteriormente diagnosticados como CRION estão sendo reclassificados como MOGAD ou NMOSD. Espera-se que a redefinição dos conceitos de doença com base no perfil de anticorpos progrida.
Jeyakumar N, Lerch M, Dale RC, Ramanathan S. MOG antibody-associated optic neuritis. Eye (London, England). 2024;38(12):2289-2301. doi:10.1038/s41433-024-03108-y. PMID:38783085; PMCID:PMC11306565.
Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. The Lancet. Neurology. 2023;22(3):268-282. doi:10.1016/S1474-4422(22)00431-8. PMID:36706773.
Varley JA, Champsas D, Prossor T, et al. Validation of the 2023 International Diagnostic Criteria for MOGAD in a Selected Cohort of Adults and Children. Neurology. 2024.
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment: A Tale of Two Central Nervous System Autoimmune Inflammatory Disorders. Neurol Clin. 2024;42(1):77-114. doi:10.1016/j.ncl.2023.06.009. PMID:37980124; PMCID:PMC10658081.
Petzold A, Woodhall M, Khaleeli Z, Tobin WO, Pittock SJ, Weinshenker BG, et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein antibodies in immune-mediated optic neuritis at long-term follow-up. Journal of neurology, neurosurgery, and psychiatry. 2019;90(9):1021-1026. doi:10.1136/jnnp-2019-320493. PMID:31118222.
Lee HJ, Kim B, Waters P, Woodhall M, Irani S, Ahn S, et al. Chronic relapsing inflammatory optic neuropathy (CRION): a manifestation of myelin oligodendrocyte glycoprotein antibodies. Journal of neuroinflammation. 2018;15(1):302. doi:10.1186/s12974-018-1335-x. PMID:30382857; PMCID:PMC6208174.
Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT. Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. Journal of neurology, neurosurgery, and psychiatry. 2010;81(1):109-11. doi:10.1136/jnnp.2008.146894. PMID:20019228.
McElhinney K, Rhatigan M, Tsvetanova Z, O’Keane C, Logan P. Beware the Retrobulbar Optic Neuritis Diagnosis. Case reports in ophthalmology. 2022;13(2):453-458. doi:10.1159/000524685. PMID:35950025; PMCID:PMC9247490.
Jossy A, Jacob N, Sarkar S, Gokhale T, Kaliaperumal S, Deb AK. COVID-19-associated optic neuritis - A case series and review of literature. Indian journal of ophthalmology. 2022;70(1):310-316. doi:10.4103/ijo.IJO_2235_21. PMID:34937266; PMCID:PMC8917537.
Bhatti MT, Gilbert AL, Watson G, et al. Shot in the dark. Surv Ophthalmol. 2023;68:821-829.
Francesca Bosello, Damiano Marastoni, Francesca Benedetta Pizzini, Chiara Zaffalon, Andrea Zuliani, Giulia Turri, Sara Mariotto, Erika Bonacci, et al. Atypical myelin oligodendrocyte glycoprotein antibody–associated optic neuritis and acute demyelinating polyneuropathy after SARS-CoV-2 infection: Case report and literature review. Journal of Neuroimmunology. 2023;375:578011. doi:10.1016/j.jneuroim.2022.578011.
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. Journal of neuroinflammation. 2016;13(1):280. doi:10.1186/s12974-016-0718-0. PMID:27793206; PMCID:PMC5086042.
Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. Journal of neurology, neurosurgery, and psychiatry. 2018;89(2):127-137. doi:10.1136/jnnp-2017-316880. PMID:29142145; PMCID:PMC5800335.
Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. MOG antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8-15.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.