Optikusneuritis ist eine Erkrankung, bei der der Sehnerv aus irgendeinem Grund entzündet ist und die Sehfunktion nachlässt. Nach Ausschluss infektiöser und nichtinfektiöser (toxischer, hereditärer, kompressiver) Optikusneuropathien sind die meisten Fälle idiopathisch, vermutlich aufgrund eines autoimmunen Mechanismus.
Das bevorzugte Alter ist 15–45 Jahre, häufiger bei Frauen, und es manifestiert sich als monokulare akute Optikusneuropathie. Die Sehstörung schreitet über einige Tage bis zwei Wochen fort und zeigt dann innerhalb von fünf Wochen eine Erholungstendenz.
Die jährliche Inzidenz in Japan beträgt 1,6 pro 100.000 Erwachsene. Das bevorzugte Alter ist 15–45 Jahre, Frauen machen etwa 70% aus. Die weltweite Prävalenz wird auf etwa 1–5 pro 100.000 Personen geschätzt.
Der Zusammenhang mit MS ist sehr eng: Die kumulative Wahrscheinlichkeit, innerhalb von 15 Jahren nach einer idiopathischen Optikusneuritis eine MS zu entwickeln, beträgt 50 %. Liegen im initialen MRT des Gehirns keine demyelinisierenden Läsionen vor, beträgt die Konversionsrate nur 25 %, bei mindestens einer Läsion steigt sie auf 78 %.
Bei Kindern liegt das bevorzugte Alter bei 9–10 Jahren. Je jünger das Kind, desto häufiger treten beidseitige und schwere Sehstörungen auf, insbesondere unter 5 Jahren. Die Konversionsrate zur pädiatrischen MS wird von Adachi et al. mit etwa 30 % und von Mizota et al. mit 16 % angegeben.
Die jährliche Inzidenz von MOGAD wird auf 1,6 bis 4,8 pro 1 Million Menschen geschätzt. 1)
QWerde ich nach einer Optikusneuritis in Zukunft an Multipler Sklerose erkranken?
A
Etwa 50 % der idiopathischen Optikusneuritiden entwickeln sich innerhalb von 15 Jahren zu einer MS. Liegen jedoch im initialen MRT des Gehirns keine demyelinisierenden Läsionen vor, beträgt die Konversionsrate nur 25 %. Nach der Diagnose sind eine Risikobewertung mittels MRT des Gehirns und eine Nachsorge in Zusammenarbeit mit einem Neurologen wichtig.
Wikimedia Commons. File:Optic_Neuritis.png. License: CC BY-SA.
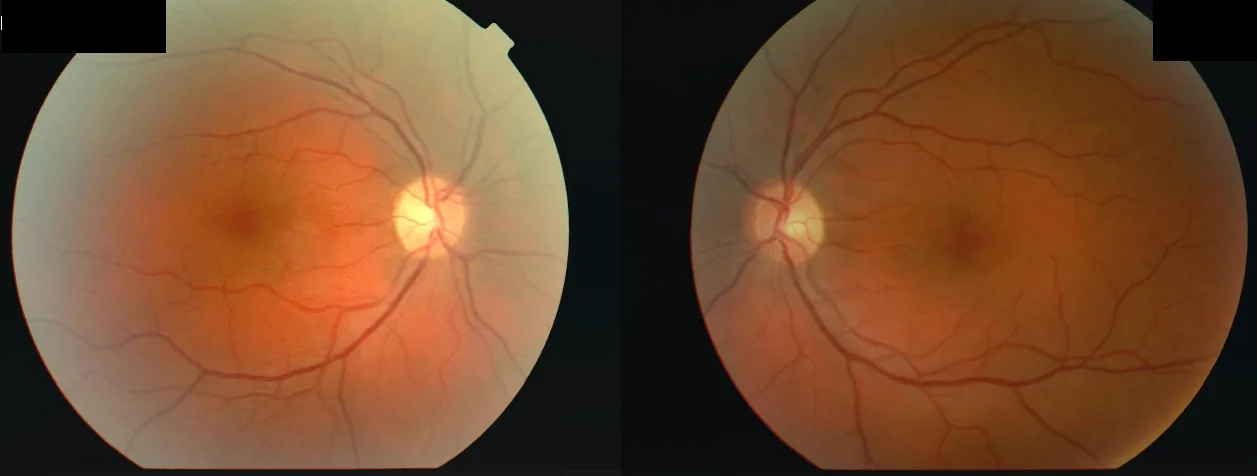
Fundusbilder beider Augen einer 34-jährigen Frau, die nach einer fieberhaften Erkrankung eine Sehverschlechterung am rechten Auge (OD 2/60) und dann am linken Auge (OS 6/24) entwickelte, mit beidseitigem Zentralskotom und einem relativen afferenten Pupillendefekt (RAPD) am rechten Auge. Dies entspricht dem „Zentralskotom“, das im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt wird.
Der Beginn ist akut, mit folgenden charakteristischen Symptomen:
Akute einseitige Sehverschlechterung: Fortschreiten über einige Tage bis zu 2 Wochen. Die Sehfunktion ist etwa eine Woche nach Beginn am schlechtesten.
Augenschmerzen oder Schmerzen bei Augenbewegungen: In Japan bei etwa 50–60 % der Fälle. Kann der Sehverschlechterung um einige Tage vorausgehen.
Gesichtsfeldausfälle: Häufig Zentralskotom oder zentrozökales Skotom, aber auch horizontale Hemianopsie möglich. Diffuse Ausfälle sind ebenfalls häufig.
Farbsehstörungen: Verminderte Sättigung von Rot, charakteristisch.
Uhthoff-Phänomen: Vorübergehende Sehverschlechterung oder Verschwommensehen nach Erhöhung der Körpertemperatur (Baden, Sport). Tritt innerhalb weniger Minuten auf und verschwindet innerhalb einer Stunde.
Phosphene: Wahrnehmung von Lichtblitzen
Pulfrich-Phänomen: Veränderung der Tiefenwahrnehmung bewegter Objekte
Relativer afferenter Pupillendefekt (RAPD): Wichtigster Befund. Positiv bei einseitigem oder asymmetrischem Befall. Bei Optikusneuritis bereits bei leichter Funktionsstörung abnorm. Vor Pupillenerweiterung mit Vorsatzlinse und Spaltlampe prüfen
Ausmaß der Sehstörung: Laut ONTT haben 10 % einen Visus ≥ 1,0, 25 % zwischen 0,5 und 1,0, 29 % zwischen 0,1 und 0,5 und 36 % darunter
Fundusbefunde: Bei anteriorer Optikusneuritis Rötung und Schwellung der Papille, in der Fluoreszenzangiographie Hyperfluoreszenz. Bei retrobulbärer Optikusneuritis initial normal, nach 4–6 Wochen Abblassung
Bei der kindlichen Optikusneuritis tritt häufiger eine typische Papillitis mit Rötung und Schwellung der Papille auf als die bei Erwachsenen beobachtete retrobulbäre Neuritis. Die Entzündung beschränkt sich nicht nur auf die Papille, sondern erstreckt sich über die gesamte Länge des Sehnervs. In 50–75 % der Fälle ist sie beidseitig, und die anteriore Optikusneuritis macht 50–75 % der Fälle aus.
QWas ist das Uhthoff-Zeichen?
A
Das Uhthoff-Zeichen ist ein Phänomen vorübergehender Sehstörung bei Anstieg der Körpertemperatur. Nach einem Bad oder Sport bemerkt der Patient eine Verschlechterung der Sehschärfe oder verschwommenes Sehen. Es tritt nach wenigen Minuten auf und verschwindet innerhalb einer Stunde. Es ist bei mit MS assoziierter Optikusneuritis bekannt, wurde aber auch bei anderen Optikusneuropathien berichtet und ist nicht spezifisch für MS.
QWie unterscheidet sich die Anti-AQP4-Antikörper-positive Optikusneuritis von der typischen Optikusneuritis?
A
Die Anti-AQP4-Antikörper-positive Optikusneuritis ist eine therapierefraktäre Form, die etwa 10 % der idiopathischen Optikusneuritiden ausmacht. Im Vergleich zur typischen Optikusneuritis ist die Sehschärfe bei Beginn schlechter, das Ansprechen auf Steroidtherapie geringer und die Rezidivrate höher. Sie unterscheidet sich auch durch die Tendenz zur beidseitigen Ausbreitung. Für die Diagnose ist die Messung der Serum-Anti-AQP4-Antikörper obligatorisch.
Bei idiopathischen Fällen, bei denen ein autoimmuner Mechanismus vermutet wird, infiltrieren Entzündungszellen wie Mikroglia den Sehnerv und verursachen eine Entzündung. Wiederholte Entzündungen führen zu einer Akkumulation von Schäden an den Nervenfasern, was zu einer Optikusatrophie führt.
Es wurde die Hypothese aufgestellt, dass eine Virusinfektion eine Autoimmunreaktion auslöst, und bei einigen Patienten werden im Liquor Antikörper gegen Masern, Windpocken, Influenza usw. nachgewiesen. Bei Kindern ist ein Zusammenhang mit ADEM (akute disseminierte Enzephalomyelitis) mit Fieber und Kopfschmerzen bekannt.
Geschlecht: häufiger bei Frauen (Verhältnis Männer zu Frauen etwa 1:2 bis 1:3)
Alter: tritt vor allem im Alter von 20–45 Jahren auf
Prodromalsymptome: kann einer grippeähnlichen Erkrankung vorausgehen
MS-Patienten: bis zu 75 % erleben mindestens einmal im Leben eine ON. Bei Autopsien werden bei bis zu 90 % Läsionen des Sehnervs festgestellt.
Als Auslöser für MOGAD wurden Impfungen und Infektionen berichtet, und es werden Mechanismen der Immuntoleranzstörung wie Bystander-Aktivierung vermutet. 1)
Es gibt Berichte über ON nach COVID-19-Infektion oder Impfung. Die mediane Zeit von der Impfung bis zum ON-Beginn beträgt 18 Tage, und 14 von 55 Fällen waren MOG-IgG-positiv. 11)
Die Optikusneuritis ist eine klinische Diagnose. Sie wird durch die Kombination von akuter einseitiger Sehverschlechterung, Schmerzen bei Augenbewegungen, positivem RAPD und Gesichtsfeldausfall diagnostiziert.
Orbitale MRT : koronale fettunterdrückte STIR und kontrastverstärkte T1-gewichtete Aufnahmen. Beurteilung von Vergrößerung, Hyperintensität und Kontrastmittelaufnahme des Sehnervs
Kopf-MRT FLAIR : Beurteilung auf demyelinisierende Läsionen im Gehirn. Unverzichtbar für die Beurteilung einer MS-Begleitung und des zukünftigen MS-Risikos
Typische ON und atypische ON unterscheiden sich in den MRT-Befunden wie folgt.
Messung der peripapillären retinalen Nervenfaserschichtdicke (pRNFL). In der akuten Phase zeigt sich eine pRNFL-Verdickung (MOG-ON Median 164 μm vs. MS-ON 103 μm). In der chronischen Phase kommt es zu einer pRNFL-Ausdünnung, und die Anzahl der Rezidive korreliert mit der pRNFL-Abnahme. Unterhalb eines pRNFL-Schwellenwerts von 50 μm verschlechtert sich die mittlere Abweichung des Gesichtsfelds signifikant. 1)
Bei atypischem Verlauf werden folgende Werte gemessen.
Anti-AQP4-Antikörper: 2013 in die Krankenversicherung aufgenommen. Bei Steroidresistenz oder wiederholten Rückfällen ist eine frühzeitige Beurteilung wichtig.
Anti-MOG-Antikörper (MOG-IgG): Serumtest mittels Live-Cell-Based Assay wird empfohlen. Fixierte CBA haben eine geringere Sensitivität und Spezifität.
Nach den internationalen MOGAD-Diagnosekriterien von 2023 wird die Diagnose durch positiven CBA + klinischen Kernphänotyp + Ausschluss von Alternativdiagnosen bestätigt. Bei niedrigem Titer ist mindestens ein unterstützendes klinisches/MRT-Merkmal erforderlich 2).
Beurteilen, ob die Optikusneuritis das erste Symptom einer MS sein könnte. Die Diagnose erfolgt durch den Nachweis einer zeitlichen und räumlichen Dissemination entzündlicher demyelinisierender Läsionen des Zentralnervensystems und durch den Ausschluss anderer Erkrankungen. Eine räumliche Dissemination kann nachgewiesen werden, wenn T2-hyperintense Läsionen in mindestens zwei der vier Regionen (periventrikulär, subkortikal, infratentoriell und Rückenmark) vorliegen.
Es wurde berichtet, dass ein Fall, der als retrobulbäre Neuritis diagnostiziert wurde, tatsächlich eine kompressive Optikusneuropathie aufgrund eines orbitalen Lymphoms war. Bei atypischen Merkmalen sollten nicht leichtfertig Steroide verabreicht werden, sondern eine gründliche Untersuchung erfolgen. 9)
In der Regel sind die bei Erwachsenen durchgeführten Untersuchungen wie FA (Fluoreszenzangiographie), CFF und dynamische Perimetrie für Kinder nicht geeignet. Die Diagnose erfolgt anhand von Fundusbefunden und MRT des Kopfes.
QWas sind die entscheidenden Punkte im MRT, um typische von atypischer Optikusneuritis zu unterscheiden?
A
Bei typischer ON zeigt sich nur ein kurzes Segment des Sehnervs mit Kontrastmittelanreicherung. Bei atypischer ON hingegen zeigt sich eine lange Kontrastmittelanreicherung des Sehnervs (mehr als die Hälfte der Länge), eine Ausdehnung nach hinten (Chiasma opticum, Tractus opticus) und eine Kontrastmittelanreicherung der Sehnervenscheide. Diese MRT-Befunde sind direkt mit der Differenzialdiagnose der zugrunde liegenden Erkrankung verbunden.
Die visuelle Prognose der idiopathischen Optikusneuritis ist gut. Bei über 90 % der Optikusneuritiden bessert sich die Sehschärfe durch Beobachtung oder systemische Steroidgabe. Nach den Langzeitergebnissen der ONTT beträgt die Sehschärfe ein Jahr nach Beginn bei 93 % 0,5 oder mehr und bei über 70 % 1,0 oder mehr.
Bei idiopathischer Optikusneuritis mit relativ guter korrigierter Sehschärfe kann eine Beobachtung unter oraler Gabe von Mecobalamin 1.500 μg/Tag (nicht erstattungsfähig) erfolgen.
Typische ON
Erste Wahl: Steroid-Pulstherapie (Methylprednisolon 1.000 mg/Tag i.v. Infusion über 3 Tage). Nicht erstattungsfähig.
Nach dem Puls: Beginn mit oralem Prednisolon 0,5 mg/kg/Tag, dann alle 3–4 Tage um 5–10 mg reduzieren.
Bei fehlendem Ansprechen auf erste Gabe: Zweiten Puls nach einem Intervall von 4–5 Tagen durchführen.
Wirkung: Verkürzt die Erholungszeit, aber es gibt keinen signifikanten Unterschied in der endgültigen Sehschärfe nach einem Jahr.
Aktive Indikation : Bilaterale Erkrankung, schwere Sehbehinderung, einziges funktionelles Auge, Rezidiv, Demyelinisierungsherde im MRT, oder starker Wunsch des Patienten nach rascher Besserung.
Anti-AQP4-Antikörper-positive Optikusneuritis
Akutphase : Steroid-Pulstherapie (erste Wahl).
Bei Unwirksamkeit : Nach 3-4 Tagen erneuter Puls → weiterhin unwirksam → Plasmapherese erwägen.
Akutphase : Steroid-Pulstherapie ist hochwirksam. Vollständige Erholung 50%, teilweise 44%. 13)
Rezidivneigung : Starke Steroidabhängigkeit, 70% Rezidive bei Reduktion von oralem Prednisolon (insbesondere <10 mg/Tag oder innerhalb von 2 Monaten nach Absetzen). 14)
Erhaltungstherapie : IVIg (≥1 g/kg/4 Wochen reduziert Rezidive signifikant). 15)Rituximab kann weniger wirksam sein als bei AQP4-ON.
Behandlungsbeginn : In der Regel nach dem zweiten Schub (da >50% der Fälle monophasisch sind).
Die orale Prednisolon-Monotherapie in Standarddosis (1 mg/kg/Tag) wird nicht empfohlen, da sie in der ONTT-Studie eine höhere Rückfallrate als Placebo oder intravenöse Steroide aufwies.
Führen Sie eine aggressive Steroidtherapie durch. Bei schlechtem Ansprechen auf den Steroidpuls sind hochdosierte intravenöse Immunglobuline und Plasmaaustausch Optionen.
Bei MS-assoziierten Fällen nach Sehverbesserung eine Behandlung zur Rückfallprävention in Betracht ziehen. In Zusammenarbeit mit einem Neurologen krankheitsmodifizierende Medikamente (Interferon beta, Glatirameracetat, Fingolimod, Natalizumab usw.) erwägen.
QBeeinflusst die Steroidbehandlung die endgültige Seherholung?
A
Laut den Ergebnissen der ONTT beschleunigt die Steroid-Pulstherapie die Erholungsgeschwindigkeit, hat aber nach einem Jahr keinen signifikanten Einfluss auf die endgültige Sehschärfe. Bei idiopathischer Optikusneuritis erholen sich auch ohne Behandlung 93 % der Patienten auf eine Sehschärfe von 0,5 oder besser. Anti-AQP4-Antikörper-positive Fälle sind jedoch steroidresistent und erfordern eine frühzeitige aggressive Behandlung einschließlich Plasmaaustausch.
QWie lange muss die Immunsuppression fortgesetzt werden?
A
Da die CRION eine Erkrankung ist, die bei Therapieabbruch rezidiviert, ist auch nach Symptomremission oft eine langfristige Immunsuppression erforderlich. Nach Bestimmung der minimal wirksamen Steroiddosis werden nichtsteroidale Immunsuppressiva hinzugefügt, um ein individuelles Behandlungsschema zu erstellen.
6. Pathophysiologie und detaillierte Entstehungsmechanismen
Bei der idiopathischen Form, bei der ein autoimmuner Mechanismus vermutet wird, infiltrieren Entzündungszellen wie Mikroglia den Sehnerv und verursachen eine Entzündung.
Immunvermittelte Zerstörung der Myelinscheide: Die Myelinscheide des Sehnervs wird durch einen Autoimmunmechanismus angegriffen. Die saltatorische Erregungsleitung wird unmöglich, was zu einer Störung der axonalen Impulsleitung führt.
Axonale Degeneration: Nach der Zerstörung der Myelinscheide beginnen die Axone der retinalen Ganglienzellen zu degenerieren.
Phagozytose durch Makrophagen: Makrophagen entfernen die restliche Myelinscheide.
Gliose: Astrozyten proliferieren und bilden eine Glianarbe. Der Begriff „Sklerose“ bei Multipler Sklerose leitet sich hiervon ab.
Wiederholte Entzündungen des Sehnervs führen zu kumulativen Schäden an den Nervenfasern, was zu einer Optikusatrophie führt.
Anti-AQP4-Antikörper binden an Komplement und greifen Astrozyten, die Gliazellen im Sehnerv, an, was zur Erkrankung führt. Astrozyten im Sehnerv und Chiasma opticum exprimieren viel AQP4 und sind daher anfällige Ziele.
Mechanismus der Anti-MOG-Antikörper-positiven Optikusneuritis
MOG-IgG (hauptsächlich IgG1-Subklasse) zielt auf MOG auf der Oberfläche der ZNS-Myelinsscheide ab. Die Aktivierung des Komplementwegs und die antikörperabhängige zelluläre Phagozytose sind an der Demyelinisierung beteiligt. Die Komplementaktivierung ist jedoch im Vergleich zu AQP4-IgG schwächer. Bei MOGAD überwiegen CD4-positive T-Zellen und Makrophagen, während bei MS CD8-positive T-Zellen vorherrschen. 1)4)
Obwohl MOG in der Netzhaut nicht exprimiert wird, kommt es bei MOG-ON zu einer Schädigung der retinalen Ganglienzellen. Als Mechanismus werden Glutamat-Exzitotoxizität und die Anfälligkeit der Blut-Hirn-Schranke am Sehnervenkopf vorgeschlagen. 1)
Auch der Mechanismus, dass IL-6 die Permeabilität der Blut-Hirn-Schranke erhöht und die Plasmablastendifferenzierung fördert, wird untersucht.
CRION wurde ursprünglich als ein Syndrom vorgeschlagen, das steroidresponsive und rezidivierende Optikusneuritiden umfasst. 5) Spätere Antikörpertests in CRION-Kohorten zeigten, dass bis zu 22 % AQP4-IgG-positiv und bis zu 25 % MOG-IgG-positiv waren. 6)7)8) Daher wird CRION als syndromale Diagnose betrachtet, die eine heterogene Gruppe von Ätiologien einschließlich MOG-Antikörper-assoziierter Optikusneuritis und AQP4-Antikörper-assoziierter Optikusneuritis umfasst.
Es wird die Hypothese aufgestellt, dass eine Virusinfektion eine Autoimmunreaktion auslöst. Es gibt Berichte über den Nachweis von DNA des Varizella-Zoster-Virus und des Herpes-simplex-Virus im Liquor einiger ON-Patienten. Bei MOGAD wird eine Störung der Immuntoleranz (Bystander-Aktivierung, molekulare Mimikry) durch Impfung oder Infektion angenommen. 1)
Phase-3-RCTs zu Satralizumab (NCT05271409) und Rozanolixizumab (NCT05063162) laufen. Bei Off-Label-Anwendung von Tocilizumab (IL-6-Rezeptor-Antikörper) wurde eine Rückfallprävention von bis zu 29 Monaten berichtet. 1)
Es wurden Fälle von demyelinisierender Optikusneuritis nach COVID-19-Infektion berichtet. Jossy et al. (2022) berichteten über Fälle von Optikusneuritis in der Erholungsphase der Infektion, bei denen alle nach einer Steroid-Pulstherapie die Sehkraft wiedererlangten. 10)
Es häufen sich Berichte über ON nach COVID-19-Impfung, mit einem medianen Intervall von 18 Tagen zwischen Impfung und ON-Beginn. Von 55 Fällen waren 14 MOG-IgG-positiv, und es gab keine AQP4-IgG-positiven Fälle. 11) Es wurden auch Fälle von MOG-Antikörper-assoziierter Optikusneuritis nach SARS-CoV-2-Infektion berichtet. 12)
Mit der Verbreitung von MOG-IgG- und AQP4-IgG-Antikörpertests werden einige Fälle, die zuvor als CRION diagnostiziert wurden, in MOGAD oder NMOSD umklassifiziert. Es wird erwartet, dass in Zukunft eine Neudefinition der Krankheitskonzepte auf der Grundlage des Antikörperprofils voranschreitet.
Jeyakumar N, Lerch M, Dale RC, Ramanathan S. MOG antibody-associated optic neuritis. Eye (London, England). 2024;38(12):2289-2301. doi:10.1038/s41433-024-03108-y. PMID:38783085; PMCID:PMC11306565.
Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. The Lancet. Neurology. 2023;22(3):268-282. doi:10.1016/S1474-4422(22)00431-8. PMID:36706773.
Varley JA, Champsas D, Prossor T, et al. Validation of the 2023 International Diagnostic Criteria for MOGAD in a Selected Cohort of Adults and Children. Neurology. 2024.
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment: A Tale of Two Central Nervous System Autoimmune Inflammatory Disorders. Neurol Clin. 2024;42(1):77-114. doi:10.1016/j.ncl.2023.06.009. PMID:37980124; PMCID:PMC10658081.
Petzold A, Woodhall M, Khaleeli Z, Tobin WO, Pittock SJ, Weinshenker BG, et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein antibodies in immune-mediated optic neuritis at long-term follow-up. Journal of neurology, neurosurgery, and psychiatry. 2019;90(9):1021-1026. doi:10.1136/jnnp-2019-320493. PMID:31118222.
Lee HJ, Kim B, Waters P, Woodhall M, Irani S, Ahn S, et al. Chronic relapsing inflammatory optic neuropathy (CRION): a manifestation of myelin oligodendrocyte glycoprotein antibodies. Journal of neuroinflammation. 2018;15(1):302. doi:10.1186/s12974-018-1335-x. PMID:30382857; PMCID:PMC6208174.
Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT. Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. Journal of neurology, neurosurgery, and psychiatry. 2010;81(1):109-11. doi:10.1136/jnnp.2008.146894. PMID:20019228.
McElhinney K, Rhatigan M, Tsvetanova Z, O’Keane C, Logan P. Beware the Retrobulbar Optic Neuritis Diagnosis. Case reports in ophthalmology. 2022;13(2):453-458. doi:10.1159/000524685. PMID:35950025; PMCID:PMC9247490.
Jossy A, Jacob N, Sarkar S, Gokhale T, Kaliaperumal S, Deb AK. COVID-19-associated optic neuritis - A case series and review of literature. Indian journal of ophthalmology. 2022;70(1):310-316. doi:10.4103/ijo.IJO_2235_21. PMID:34937266; PMCID:PMC8917537.
Bhatti MT, Gilbert AL, Watson G, et al. Shot in the dark. Surv Ophthalmol. 2023;68:821-829.
Francesca Bosello, Damiano Marastoni, Francesca Benedetta Pizzini, Chiara Zaffalon, Andrea Zuliani, Giulia Turri, Sara Mariotto, Erika Bonacci, et al. Atypical myelin oligodendrocyte glycoprotein antibody–associated optic neuritis and acute demyelinating polyneuropathy after SARS-CoV-2 infection: Case report and literature review. Journal of Neuroimmunology. 2023;375:578011. doi:10.1016/j.jneuroim.2022.578011.
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. Journal of neuroinflammation. 2016;13(1):280. doi:10.1186/s12974-016-0718-0. PMID:27793206; PMCID:PMC5086042.
Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. Journal of neurology, neurosurgery, and psychiatry. 2018;89(2):127-137. doi:10.1136/jnnp-2017-316880. PMID:29142145; PMCID:PMC5800335.
Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. MOG antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8-15.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.