Optic neuritis is a disease in which the optic nerve becomes inflamed for some reason, leading to decreased visual function. After excluding infectious and non-infectious (toxic, hereditary, compressive) optic neuropathies, the majority are idiopathic, thought to be caused by autoimmune mechanisms.
Peak age of onset is 15–45 years, more common in women, and presents as acute monocular optic neuropathy. Visual dysfunction progresses over several days to 2 weeks, then shows a tendency to recover within 5 weeks.
Anterior optic neuritis (optic disc inflammation): Accompanied by optic disc swelling. Seen in about 50% in Japan (higher than 35% in Western countries).
Retrobulbar optic neuritis: The fundus appears normal in the acute phase. Optic disc pallor appears after 4–6 weeks.
The annual incidence in Japan is 1.6 per 100,000 adults. Peak age is 15–45 years, with women accounting for about 70%. The global prevalence is estimated at 1–5 per 100,000 people.
The association with MS is very strong, with a cumulative probability of conversion to MS of 50% within 15 years after onset of idiopathic optic neuritis. If there are no demyelinating lesions on brain MRI at first onset, the conversion rate remains at 25%, but if there is one or more lesions, it reaches 78%.
In children, the peak age of onset is 9–10 years, and younger children tend to present with bilateral and severe visual impairment, especially those under 5 years old. The conversion rate to pediatric MS has been reported as approximately 30% by Adachi et al. and 16% by Mizota et al.
The annual incidence of MOGAD is estimated to be 1.6–4.8 per million people. 1)
QIf I develop optic neuritis, will I get multiple sclerosis in the future?
A
Approximately 50% of idiopathic optic neuritis cases convert to MS within 15 years. However, if there are no demyelinating lesions on brain MRI at first onset, the conversion rate remains at 25%. After onset, risk assessment using brain MRI and follow-up in collaboration with a neurologist is important.
Wikimedia Commons. File:Optic_Neuritis.png. License: CC BY-SA.
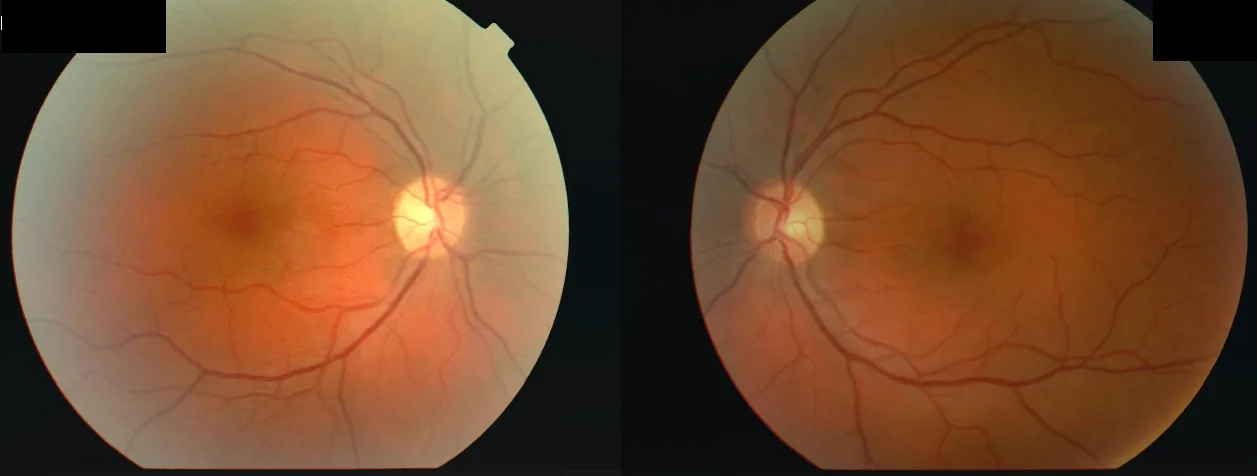
Fundus photographs of a 34-year-old woman who developed decreased vision in the right eye (OD 2/60) followed by the left eye (OS 6/24) after a febrile illness, with central scotomas in both eyes and a relative afferent pupillary defect (RAPD) in the right eye. This corresponds to the “central scotoma” discussed in the section “2. Main Symptoms and Clinical Findings”.
It presents with an acute onset pattern, and the following symptoms are characteristic:
Acute unilateral visual loss: Progresses over several days to about 2 weeks. Visual function is worst around 1 week after onset.
Eye pain / pain on eye movement: Seen in about 50–60% of cases in Japan. May appear a few days before visual loss.
Visual field defects: Central scotoma or centrocecal scotoma are common, but altitudinal hemianopia may also occur. Diffuse defects are also frequent.
Color vision abnormality: Decreased saturation of red color is characteristic.
Uhthoff phenomenon: Transient visual loss or blurring after an increase in body temperature (e.g., hot bath, exercise). Appears within minutes and resolves within 1 hour.
Relative afferent pupillary defect (RAPD): Most important finding. Positive when unilateral or asymmetric. In optic neuritis, it may be abnormal even with mild dysfunction. Check with a condensing lens and slit lamp before dilation.
Degree of visual impairment: In ONTT, visual acuity ≥1.0 in 10%, 0.5–1.0 in 25%, 0.1–0.5 in 29%, and worse in 36%.
Fundus findings: In anterior optic neuritis, optic dischyperemia and swelling are seen, with hyperfluorescence on fluorescein angiography. In retrobulbar optic neuritis, the fundus is normal early, with pallor after 4–6 weeks.
In pediatric optic neuritis, typical papillitis with redness and swelling of the optic disc is more common than retrobulbar optic neuritis seen in adults. The inflammation extends along the entire length of the optic nerve, not just the disc. Bilateral involvement occurs in 50–75% of cases, and anterior optic neuritis type accounts for 50–75%.
QWhat is Uhthoff's sign?
A
Uhthoff’s sign is a phenomenon of transient visual disturbance associated with an increase in body temperature. Patients notice decreased vision or blurred vision after bathing or exercise. It appears within minutes and resolves within one hour. It is known in optic neuritis associated with MS but has also been reported in other optic neuropathies and is not specific to MS.
QHow does anti-AQP4 antibody-positive optic neuritis differ from typical optic neuritis?
A
Anti-AQP4 antibody-positive optic neuritis is a refractory subtype accounting for about 10% of idiopathic optic neuritis. Compared to typical optic neuritis, visual acuity at onset is worse, response to steroid therapy is poor, and recurrence is more frequent. It also tends to become bilateral. Measurement of serum anti-AQP4 antibodies is essential for diagnosis.
In idiopathic cases suspected to have an autoimmune mechanism, inflammatory cells such as microglia infiltrate the optic nerve and cause inflammation. Repeated inflammation leads to cumulative damage to nerve fibers and progression to optic atrophy.
A hypothesis has been proposed that viral infection triggers an autoimmune reaction, and viral antibodies against measles, varicella, influenza, etc., are detected in the cerebrospinal fluid of some patients. In children, an association with ADEM (acute disseminated encephalomyelitis) accompanied by fever and headache is known.
Sex: More common in women (male-to-female ratio approximately 1:2 to 1:3)
Age: Most common between 20 and 45 years old
Prodromal symptoms: May be preceded by an influenza-like illness
MS patients: Up to 75% experience at least one episode of ON in their lifetime. Autopsy studies show optic nerve lesions in up to 90%.
Vaccination and infection have been reported as triggers for MOGAD onset, and mechanisms such as bystander activation leading to breakdown of immune tolerance are suspected. 1)
There are reports of ON onset after COVID-19 infection or vaccination. The median time from vaccination to ON onset was 18 days, and 14 out of 55 cases were MOG-IgG positive. 11)
Optic neuritis is a clinical diagnosis. It is diagnosed based on a combination of acute unilateral vision loss, eye movement pain, positive RAPD, and visual field defects.
Measures peripapillary retinal nerve fiber layer (pRNFL) thickness. In the acute phase, pRNFL thickening is observed (MOG-ON median 164 μm vs MS-ON 103 μm). In the chronic phase, pRNFL thinning occurs, and the number of relapses correlates with pRNFL reduction. Visual field mean deviation is significantly worse below a pRNFL threshold of 50 μm. 1)
Anti-AQP4 antibody: Covered by insurance in 2013. Early evaluation is important in cases of steroid resistance or recurrent relapses.
Anti-MOG antibody (MOG-IgG): Serum testing by live cell-based assay is recommended. Fixed CBA has inferior sensitivity and specificity.
The 2023 international diagnostic criteria for MOGAD define a definite diagnosis as positive CBA plus core clinical phenotype plus exclusion of alternative diagnoses. In cases of low titer, at least one supportive clinical/MRI feature is required 2).
Validation of these criteria: sensitivity 96.5%, specificity 98.9%, accuracy 98.5% 3).
Evaluate the possibility that optic neuritis is the initial symptom of MS. Diagnosis is made by demonstrating dissemination in time and space of inflammatory demyelinating lesions in the central nervous system and excluding other diseases. Dissemination in space can be demonstrated if T2 hyperintense lesions are present in at least two of four areas: periventricular, cortical/juxtacortical, infratentorial, and spinal cord.
Accompanied by fever, headache, altered consciousness
There have been reports of cases diagnosed with retrobulbar optic neuritis that were actually compressive optic neuropathy due to orbital lymphoma. If atypical features are present, steroids should not be administered casually, and a thorough examination should be performed. 9)
Usually, FA (fluorescein angiography), CFF, and kinetic perimetry performed in adults are not suitable for children; diagnosis is made by fundus findings and head MRI.
QWhat are the MRI points to distinguish typical from atypical optic neuritis?
A
In typical ON, contrast enhancement is seen only in a short segment of the optic nerve. In contrast, atypical ON shows findings such as long-segment contrast enhancement (more than half the length), posterior extension (to the optic chiasm and optic tract), and enhancement of the optic nerve sheath. These MRI findings are directly linked to the differential diagnosis of the underlying disease.
The visual prognosis of idiopathic optic neuritis is good. In more than 90% of optic neuritis cases, visual acuity improves with observation or systemic steroid administration. According to the long-term results of the ONTT, visual acuity at 1 year after onset is 0.5 or better in 93% and 1.0 or better in more than 70%.
In idiopathic optic neuritis with relatively good corrected visual acuity, oral mecobalamin 1,500 μg/day (off-label use) may be used for observation.
Typical ON
First-line treatment: Steroid pulse therapy (methylprednisolone 1,000 mg/day IV drip infusion for 3 days). Off-label use.
After pulse: Start oral prednisolone at 0.5 mg/kg/day, then taper by 5–10 mg every 3–4 days.
If first pulse ineffective: Administer a second pulse after an interval of 4–5 days.
Effect: Shortens the recovery period, but there is no significant difference in final visual acuity at 1 year after onset.
Active indication: Bilateral onset, severe visual dysfunction, only functional eye, recurrent cases, MRI demyelinating plaques present, or patient strongly desires early improvement.
Anti-AQP4 antibody-positive ON
Acute phase: Steroid pulse therapy (first-line).
If ineffective: Repeat pulse after 3–4 days → if still ineffective → consider plasma exchange.
Plasma exchange: Simple plasma exchange > double filtration plasmapheresis > immunoadsorption (in order of efficacy). One course: 5–6 sessions.
Relapse tendency: Strong steroid dependence; 70% relapse during tapering of oral prednisolone (especially <10 mg/day or within 2 months after discontinuation). 14)
Maintenance therapy: IVIg (≥1 g/kg every 4 weeks significantly reduces relapse). 15)Rituximab may be less effective than for AQP4-ON.
Timing of treatment initiation: Usually after the second attack (because >50% are monophasic).
Standard-dose (1 mg/kg/day) oral prednisolone monotherapy is not recommended because it was associated with a higher relapse rate compared to placebo or intravenous steroids in the ONTT.
Aggressive steroid therapy is performed. If response to steroid pulse is poor, high-dose intravenous immunoglobulin or plasma exchange therapy are options.
In cases with MS, consider treatment for relapse prevention after visual improvement. In collaboration with a neurologist, consider disease-modifying drugs (interferon beta, glatiramer acetate, fingolimod, natalizumab, etc.).
QDoes steroid treatment affect final visual recovery?
A
According to the ONTT results, steroid pulse therapy shortens recovery time but does not significantly affect final visual acuity one year after onset. In idiopathic optic neuritis, 93% of untreated patients recover to visual acuity of 0.5 or better. However, anti-AQP4 antibody-positive cases are steroid-resistant, requiring aggressive treatment including early plasma exchange.
QHow long should immunosuppressive therapy be continued?
A
Since CRION is a disease that relapses upon treatment discontinuation, long-term immunosuppressive therapy is often necessary even after symptom remission. After identifying the minimum effective dose of steroids, non-steroidal immunosuppressants are added, and the treatment regimen is individually designed.
In idiopathic cases suspected to involve autoimmune mechanisms, inflammatory cells such as microglia infiltrate the optic nerve and cause inflammation.
Immune-mediated demyelination: The myelin sheath of the optic nerve is attacked by autoimmune mechanisms. Saltatory conduction becomes impossible, leading to impaired axonal impulse conduction.
Axonal degeneration: After demyelination, axons of retinal ganglion cells begin to degenerate.
Phagocytosis by macrophages: Macrophages remove residual myelin.
Gliosis: Astrocytes proliferate and form glial scars. The term “sclerosis” in multiple sclerosis originates from this process.
Repeated episodes of optic nerve inflammation cause cumulative damage to nerve fibers, leading to optic atrophy.
Mechanism of Anti-AQP4 Antibody-Positive Optic Neuritis
Anti-AQP4 antibodies bind to complement and attack astrocytes, the glial cells in the optic nerve, leading to disease onset. Astrocytes in the optic nerve and optic chiasm highly express AQP4, making them susceptible targets.
MOG-IgG (mainly IgG1 subclass) targets MOG on the CNS myelin surface. Complement pathway activation and antibody-dependent cellular phagocytosis are involved in demyelination. However, complement activation is weaker compared to AQP4-IgG. In MOGAD, CD4-positive T cells and macrophages predominate, whereas in MS, CD8-positive T cells are predominant. 1)4)
Although MOG is not expressed in the retina, MOG-ON causes damage to retinal ganglion cells. Glutamate toxicity and vulnerability of the blood-brain barrier at the optic nerve head have been proposed as mechanisms. 1)
The mechanism by which IL-6 increases blood-brain barrier permeability and promotes plasmablast differentiation is also attracting attention.
CRION was originally proposed as a syndrome encompassing steroid-responsive and relapsing optic neuritis. 5) Subsequent antibody testing in CRION cohorts revealed that up to 22% are AQP4-IgG positive and up to 25% are MOG-IgG positive. 6)7)8) Therefore, CRION is considered a syndromic diagnosis encompassing heterogeneous etiologies including MOG antibody-associated optic neuritis and AQP4 antibody-associated optic neuritis.
The hypothesis that viral infection triggers autoimmune reactions has been proposed. There are reports of detection of varicella-zoster virus and herpes simplex virus DNA in the cerebrospinal fluid of some ON patients. In MOGAD, breakdown of immune tolerance due to vaccination or infection (bystander activation, molecular mimicry) is assumed. 1)
Phase 3 RCTs of satralizumab (NCT05271409) and rozanolixizumab (NCT05063162) are ongoing. Off-label use of tocilizumab (IL-6 receptor antibody) has been reported to prevent relapse for up to 29 months. 1)
Cases of demyelinating optic neuritis following COVID-19 infection have been reported. Jossy et al. (2022) reported cases of optic neuritis during the recovery phase of infection, and all patients recovered vision with steroid pulse therapy. 10)
Reports of ON onset after COVID-19 vaccination have accumulated, with a median interval of 18 days from vaccination to ON onset. Among 55 cases, 14 were MOG-IgG positive, and none were AQP4-IgG positive. 11) Cases of MOG antibody-associated optic neuritis after SARS-CoV-2 infection have also been reported. 12)
With the widespread use of antibody testing for MOG-IgG and AQP4-IgG, some cases previously diagnosed as CRION are being reclassified as MOGAD or NMOSD. It is expected that the disease concept will be redefined based on antibody profiles in the future.
Jeyakumar N, Lerch M, Dale RC, Ramanathan S. MOG antibody-associated optic neuritis. Eye (London, England). 2024;38(12):2289-2301. doi:10.1038/s41433-024-03108-y. PMID:38783085; PMCID:PMC11306565.
Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. The Lancet. Neurology. 2023;22(3):268-282. doi:10.1016/S1474-4422(22)00431-8. PMID:36706773.
Varley JA, Champsas D, Prossor T, et al. Validation of the 2023 International Diagnostic Criteria for MOGAD in a Selected Cohort of Adults and Children. Neurology. 2024.
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment: A Tale of Two Central Nervous System Autoimmune Inflammatory Disorders. Neurol Clin. 2024;42(1):77-114. doi:10.1016/j.ncl.2023.06.009. PMID:37980124; PMCID:PMC10658081.
Petzold A, Woodhall M, Khaleeli Z, Tobin WO, Pittock SJ, Weinshenker BG, et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein antibodies in immune-mediated optic neuritis at long-term follow-up. Journal of neurology, neurosurgery, and psychiatry. 2019;90(9):1021-1026. doi:10.1136/jnnp-2019-320493. PMID:31118222.
Lee HJ, Kim B, Waters P, Woodhall M, Irani S, Ahn S, et al. Chronic relapsing inflammatory optic neuropathy (CRION): a manifestation of myelin oligodendrocyte glycoprotein antibodies. Journal of neuroinflammation. 2018;15(1):302. doi:10.1186/s12974-018-1335-x. PMID:30382857; PMCID:PMC6208174.
Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT. Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. Journal of neurology, neurosurgery, and psychiatry. 2010;81(1):109-11. doi:10.1136/jnnp.2008.146894. PMID:20019228.
McElhinney K, Rhatigan M, Tsvetanova Z, O’Keane C, Logan P. Beware the Retrobulbar Optic Neuritis Diagnosis. Case reports in ophthalmology. 2022;13(2):453-458. doi:10.1159/000524685. PMID:35950025; PMCID:PMC9247490.
Jossy A, Jacob N, Sarkar S, Gokhale T, Kaliaperumal S, Deb AK. COVID-19-associated optic neuritis - A case series and review of literature. Indian journal of ophthalmology. 2022;70(1):310-316. doi:10.4103/ijo.IJO_2235_21. PMID:34937266; PMCID:PMC8917537.
Bhatti MT, Gilbert AL, Watson G, et al. Shot in the dark. Surv Ophthalmol. 2023;68:821-829.
Francesca Bosello, Damiano Marastoni, Francesca Benedetta Pizzini, Chiara Zaffalon, Andrea Zuliani, Giulia Turri, Sara Mariotto, Erika Bonacci, et al. Atypical myelin oligodendrocyte glycoprotein antibody–associated optic neuritis and acute demyelinating polyneuropathy after SARS-CoV-2 infection: Case report and literature review. Journal of Neuroimmunology. 2023;375:578011. doi:10.1016/j.jneuroim.2022.578011.
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. Journal of neuroinflammation. 2016;13(1):280. doi:10.1186/s12974-016-0718-0. PMID:27793206; PMCID:PMC5086042.
Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. Journal of neurology, neurosurgery, and psychiatry. 2018;89(2):127-137. doi:10.1136/jnnp-2017-316880. PMID:29142145; PMCID:PMC5800335.
Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. MOG antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8-15.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.