Systemischer Lupus erythematodes (SLE) ist eine Autoimmunerkrankung, die durch eine chronische Überaktivierung des Immunsystems zu Entzündungen in mehreren Organen des gesamten Körpers führt. Er zählt zu den fünf klassischen Kollagenosen.
Die Erkrankung tritt vor allem bei Frauen im Alter von 20 bis 30 Jahren auf. Das Verhältnis von Männern zu Frauen beträgt etwa 1:8 bis 1:9, mit einer deutlichen weiblichen Dominanz. Sie ist häufiger bei Menschen asiatischer Abstammung und seltener bei Weißen. Die Prävalenz wird auf 50–100 pro 100.000 Einwohner geschätzt.
Die Diagnosekriterien für den systemischen Lupus erythematodes (SLE) haben sich wie folgt entwickelt.
Kriterien des American College of Rheumatology (ACR) (letzte Überarbeitung 1997) : Diagnose bei Vorliegen von mindestens 4 von 11 klinischen und immunologischen Kriterien
SLICC-Kriterien (2012) : erweitert auf 17 Punkte, Diagnose bei mindestens 4 Punkten
Kriterien der European League Against Rheumatism / des American College of Rheumatology (2019) : positive antinukleäre Antikörper (ANA) obligatorisch, Diagnose bei einem gewichteten Gesamtscore von mindestens 10 Punkten
Nach den überarbeiteten Diagnosekriterien des American College of Rheumatology ist die Diagnose möglich, wenn im Verlauf mindestens 4 der folgenden 11 Punkte positiv sind: Schmetterlingserythem, diskoiden Hautausschlag, Photosensitivität, orale Ulzera, Arthritis, Serositis, Nierenbeteiligung, neurologische Störungen, Blutbildveränderungen, immunologische Anomalien, antinukleäre Antikörper.
Darüber hinaus wird der SLE in den Leitlinien zur Behandlung von Uveitis als Differenzialdiagnose der mit Kollagenosen assoziierten Uveitis geführt1). Die Beurteilung erfolgt durch Kombination von Augen- und Allgemeinsymptomen in Zusammenarbeit mit der internistischen Rheumatologie.
QSind die Augensymptome des SLE in den Diagnosekriterien enthalten?
A
Nein. Etwa 30 % der Patienten haben Augensymptome, aber diese sind nicht Teil der SLE-Diagnosekriterien.
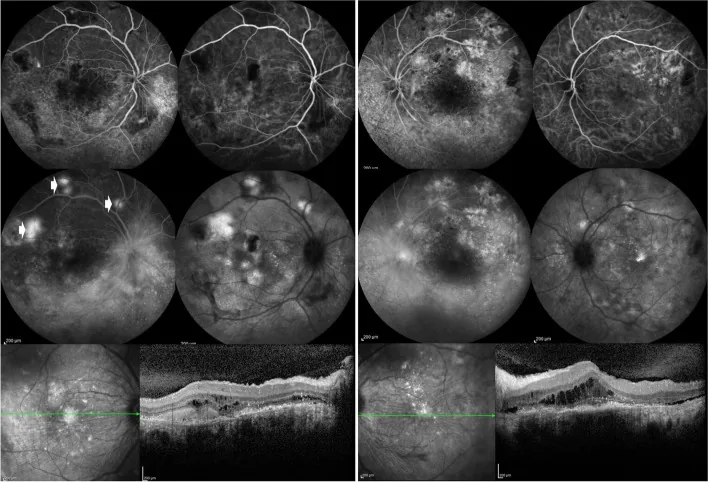
Özdal PÇ, et al. Choroidal involvement in systemic vasculitis: a systematic review. J Ophthalmic Inflamm Infect. 2022. Figure 5. PMCID: PMC8980189. License: CC BY.
Die Fluoreszenzangiographie beider Augen zeigt Leckagen an der Papille und um die Netzhautgefäße sowie fleckige Läsionen im hinteren Pol. Die OCT zeigt subretinale Flüssigkeit, was auf entzündliche Läsionen des hinteren Augenabschnitts bei SLE hinweist.
Augenbeschwerden treten bei etwa 33–50 % der Patienten auf. Die Symptome reichen von leichter Reizung bis zu schwerem Sehverlust.
Trockenheitsgefühl und Fremdkörpergefühl : die häufigsten Beschwerden. Sie sind auf ein trockenes Auge infolge eines sekundären Sjögren-Syndroms zurückzuführen. Begleitet von Brennen, verschwommenem Sehen und Verschlechterung der Symptome am Abend.
Sehverschlechterung : tritt bei Lupus-Retinopathie und Optikusneuritis auf. Sie reicht von asymptomatischen Fundusveränderungen bis zu plötzlichem Sehverlust.
Augenschmerz : Bei Optikusneuritis ist der periorbitale Schmerz charakteristisch, der sich bei Augenbewegungen verschlimmert.
Photophobie (Lichtempfindlichkeit) : tritt bei trockenem Auge und Entzündung des vorderen Augenabschnitts auf.
Farbsehstörung : Bei Optikusneuritis wird bei fast allen Patienten eine Rot-Grün-Sehstörung festgestellt.
Die Lupus-Retinopathie ist die wichtigste Veränderung des hinteren Augenabschnitts. Sie ist in der Regel beidseitig und tritt häufig in Phasen hoher Krankheitsaktivität auf.
Keratoconjunctivitis sicca : tritt bei etwa 30 % der Patienten auf. Es handelt sich um ein Tränenmangel-Syndrom.
Skleritis : Eine anteriore diffuse oder noduläre Skleritis tritt in wenigen Prozent auf. Die Ansprechbarkeit auf Steroide ist gut. Eine nekrotisierende Skleritis kann schwerwiegend sein.
Diskoidläsionen : Wenn sie die Augenlider betreffen, können sie ein narbenbedingtes Entropium oder Ektropium verursachen.
Optikusneuritis : tritt bei etwa 1 % auf. Es zeigen sich Blässe oder Ödem der Papille und ein relativer afferenter Pupillendefekt (RAPD).
Augenbewegungsstörungen : Durch Läsionen des Zentralnervensystems können der N. oculomotorius und der N. abducens betroffen sein. Tritt bei etwa 30 % auf.
QTritt Uveitis bei SLE auf?
A
Die Komplikation einer Uveitis ist überraschend selten. Bei SLE kann eine Iridozyklitis auftreten, die jedoch meist mild ist. Wenn eine Uveitis festgestellt wird, sollten andere Ursachen in Betracht gezogen werden.
Es wurden Mutationen in über 80 mit SLE assoziierten Genloci identifiziert. Ein komplexes Gleichgewicht zwischen Suszeptibilitätsgenen und protektiven Genen ist an der Krankheitsentstehung beteiligt.
Die überwältigende Häufigkeit bei Frauen lässt auf eine Beteiligung von Östrogen und anderen Hormonen schließen. Die Häufung im gebärfähigen Alter unterstützt diese Hypothese.
Die Lupus-Retinopathie ist mit einer schlechten Krankheitskontrolle verbunden. Eine Positivität für Anticardiolipin-Antikörper spielt eine wichtige Rolle bei der Entstehung von Verschlussläsionen2).
Sonstige: Anti-SS-A-/Anti-SS-B-Antikörper (Beurteilung eines sekundären Sjögren-Syndroms)
Insbesondere sind antinukleäre Antikörper in der aktiven Phase in den meisten Fällen positiv und nützlich für die Beurteilung der Krankheitsaktivität. Als Uveitis-Screening werden auch die von den Leitlinien empfohlenen Basisuntersuchungen (HLA-B27, Röntgen-Thorax, Lues-Serologie, QFT-3G, ACE, ANA) überprüft 1).
Funduskopie : Beurteilung von Cotton-Wool-Herden, Blutungen und Gefäßanomalien unter Mydriasis
Fluoreszenzangiographie (FFA) : wichtig zur Beurteilung von retinaler Vaskulitis und Gefäßverschlüssen. Erkennt Leckagen, Teleangiektasien, Verschlüsse und Mikroaneurysmen
Indocyaningrün (ICG)-Angiographie : erkennt eine in der FFA nicht sichtbare Choroidopathie als choroidale Hyperfluoreszenz
OCT-A (Optische Kohärenztomographie-Angiographie) : möglicherweise nützlich zur Überwachung struktureller Netzhautveränderungen bei subklinischer Retinopathie (Forschungsstadium)
QWie unterscheidet man die SLE-Retinopathie von der diabetischen Retinopathie?
A
Die SLE-Retinopathie ist im Vergleich zur diabetischen Retinopathie stärker okklusiv und führt leichter zu schwerer Ischämie. Eine Diabetesanamnese oder ein erhöhter HbA1c-Wert sind für die Differenzialdiagnose wichtig. In der Fluoreszenzangiographie zeigen sich bei SLE in der akuten Phase eine starke Leckage aus den Netzhautgefäßen und mikrovaskuläre Anomalien.
Die Behandlung der Augenmanifestationen des SLE basiert in erster Linie auf der Kontrolle der Grunderkrankung. Die Zusammenarbeit mit einem Rheumatologen ist unerlässlich.
Bei fortschreitendem Gefäßverschluss durch retinale Vaskulitis ist eine Antikoagulation erforderlich. Warfarin 2–5 mg/Tag, eingestellt auf einen PT-INR von 1,5–2.
Belimumab (Benlysta®) : BLyS/BAFF-Inhibitor. Zugelassen für SLE. Die BLISS-52-Studie zeigte eine signifikante Verringerung der Krankheitsaktivität3)
Anifrolumab (Saphnelo®) : Anti-IFNAR1-Antikörper (Typ-I-IFN-Rezeptor-Inhibitor). 2022 in Japan zugelassen. Verbessert signifikant die Krankheitsaktivität bei mittelschwerem bis schwerem SLE4)
Voclosporin (Calcineurininhibitor) : Indikation bei Lupusnephritis
Wird häufig zur Verringerung von SLE-Schüben eingesetzt. Ophthalmologisch ist auf Netzhauttoxizität zu achten (siehe Abschnitt « Augentoxizität von Hydroxychloroquin »).
Netzhautphotokoagulation : Bei Bestätigung einer retinalen Neovaskularisation durch Fluoreszenzangiographie sofort durchführen, um Glaskörperblutungen zu verhindern. Auch prophylaktisch bei ausgedehntem retinalem Gefäßverschluss.
Vitrektomie : Bei proliferativer Vitreoretinopathie erforderlich.
Plasmapherese : Bei schweren Fällen.
Seröse Netzhautablösung : Fluoreszenzangiographisch den Leckpunkt aus dem retinalen Pigmentepithel bestätigen und an diesem Punkt eine Netzhautphotokoagulation durchführen.
In den ersten 5 Jahren nach Behandlungsbeginn nur Basisuntersuchung. Ab dem 5. Jahr jährlich Humphrey 10-2 Gesichtsfeldtest, SD-OCT und Fundus-Autofluoreszenz empfohlen. Bei hohen Risikofaktoren früher als 5 Jahre beginnen. Bei Feststellung einer Makulopathie das Medikament absetzen.
QWie häufig sind Routineuntersuchungen bei Patienten unter Hydroxychloroquin erforderlich?
A
In den ersten 5 Jahren nach Behandlungsbeginn ist nur eine Basisuntersuchung ausreichend, aber ab dem 5. Jahr wird eine jährliche Fundusuntersuchung empfohlen. Humphrey 10-2 Gesichtsfeldtest und SD-OCT sind die wichtigsten Screening-Methoden. Bei hohen Risikofaktoren (Nierenfunktionsstörung, hohe Dosis, Langzeitanwendung) sollten die Untersuchungen früher beginnen5).
6. Pathophysiologie und detaillierte Entstehungsmechanismen
Die Pathogenese des SLE beruht auf dem Verlust der Selbsttoleranz und der Überproduktion von Autoantikörpern.
Verlust der Selbsttoleranz : Genetische und umweltbedingte Prädispositionen führen zum Verlust der Immuntoleranz gegenüber Selbstantigenen
T-Zell-Anomalien : Helfer-T-Zellen werden übermäßig aktiviert, regulatorische Immunzellen nehmen ab
B-Zell-Reifungsanomalien : B-Zellen reifen schneller, Apoptose wird unterdrückt. Plasmazellen haben eine verlängerte Lebensdauer und produzieren überschüssige Autoantikörper
Hochregulation des Typ-I-Interferon-Signalwegs : Überaktivierung von B-Zellen über BLyS/BAFF
Bildung von Immunkomplexen : Autoantikörper binden an nukleäre, intranukleäre und zytoplasmatische Selbstantigene und führen zur Freisetzung entzündlicher Zytokine
Gewebeschädigung : Chronische Entzündung, Ablagerung von Immunkomplexen und unzureichende Beseitigung apoptotischer Zellen verursachen Schäden an Geweben und Organen
Die pathologischen Befunde des SLE sind durch eine Vaskulitis mit fibrinoider Nekrose kleiner Gefäße und Kapillaren gekennzeichnet. Die fibrinoide Substanz besteht aus Fibrin, Immunkomplexen und Komplement.
Die Pathophysiologie der Lupus-Retinopathie umfasst einen dualen Mechanismus.
Immunkomplex-Vaskulitis: Die Ablagerung von Immunkomplexen am Gefäßendothel aktiviert Komplement und setzt Entzündungsmediatoren frei. Dies führt zu Nichtperfusion und Ischämie.
Thrombotischer Mechanismus: Die Entwicklung eines Antiphospholipid-Antikörper-Syndroms verursacht eine Thrombose der Netzhautgefäße. Positivität für Anticardiolipin-Antikörper ist mit okklusiver Lupus-Retinopathie assoziiert2).
Wenn sich die Nichtperfusionsbereiche ausdehnen, treten Neovaskularisationen auf, die zu einer proliferativen Vitreoretinopathie und sogar zu einem Neovaskularisationsglaukom fortschreiten können.
Neben chronischer Entzündung und Immunkomplexablagerung ist die Entwicklung eines sekundären Sjögren-Syndroms die Hauptursache. Der autoimmune Angriff auf die Tränendrüsen reduziert die Tränensekretion.
QWarum ist die SLE-Retinopathie schwerwiegender als die diabetische Retinopathie?
A
Bei der SLE-Retinopathie führt der duale Mechanismus aus Immunkomplex-Vaskulitis und Thrombose durch Antiphospholipid-Antikörper zu einem stärker okklusiven Krankheitsbild. Sie verursacht leichter eine schwere Ischämie als die diabetische Retinopathie, und die okklusiven Gefäßläsionen sind mit einer schlechteren Sehprognose verbunden.
7. Aktuelle Forschung und Zukunftsperspektiven (Berichte aus der Forschungsphase)
Die optische Kohärenztomographie-Angiographie (OCT-A) ist eine neue bildgebende Diagnosemethode, die nicht-invasiv subklinische mikrovaskuläre Netzhautveränderungen bewerten kann, die mit der herkömmlichen Fluoreszenzangiographie schwer zu erkennen sind. Ihre Anwendung zur Früherkennung und Überwachung der SLE-Retinopathie ist vielversprechend, aber ihr Nutzen ist noch nicht ausreichend untersucht.
Bei der CD19-gerichteten CAR-T-Therapie für refraktären SLE wurde bei einer kleinen Anzahl von behandlungsresistenten Patienten eine medikamentenfreie Remission berichtet6). Die langfristigen Auswirkungen auf Augenkomplikationen sind Gegenstand zukünftiger Forschung.
Anifrolumab (Typ-I-IFN-Rezeptor-Inhibitor) verbessert die Krankheitskontrolle bei aktivem SLE und könnte einen indirekten Effekt auf Augenkomplikationen haben4). Spezifische Wirksamkeitsdaten im ophthalmologischen Bereich werden weiterhin gesammelt.
Stafford-Brady FJ, Urowitz MB, Gladman DD, Easterbrook M. Lupus retinopathy. Patterns, associations, and prognosis. Arthritis Rheum. 1988;31:1105-1110.
Navarra SV, Guzmán RM, Gallacher AE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial (BLISS-52). Lancet. 2011;377:721-731. doi:10.1016/s0140-6736(10)61354-2.
Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, Bae SC, Brohawn PZ, Pineda L, Berglind A, Tummala R, TULIP-2 Trial Investigators.. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med. 2020;382(3):211-221. doi:10.1056/nejmoa1912196. PMID:31851795.
Marmor MF, Kellner U, Lai TYY, et al. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 Revision). Ophthalmology. 2016;123:1386-1394.
Mackensen A, Müller F, Mougiakakos D, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. 2022;28:2124-2132. doi:10.1038/s41591-022-02017-5.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.