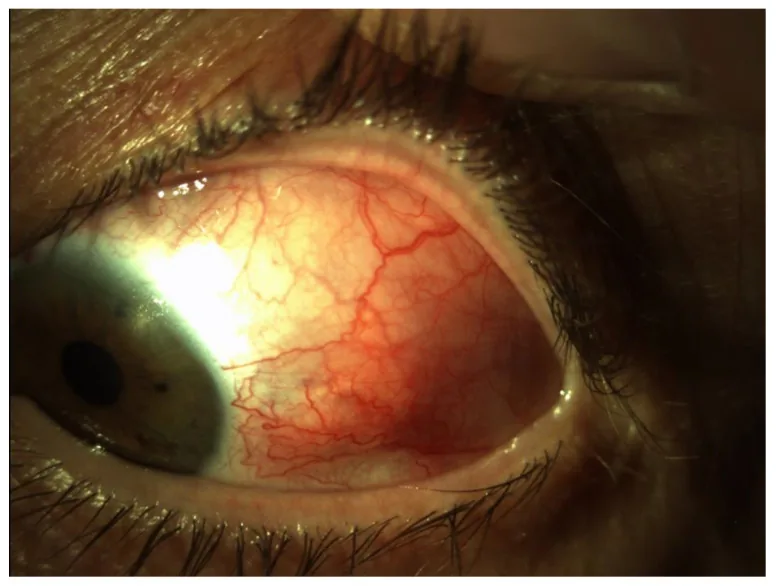
Skleritis ist eine Entzündung der tiefen Blutgefäße, wie des episkleralen und intrakleralen Gefäßplexus, die die oberflächliche Sklera bedecken, und geht mit Ödem und Zellinfiltration der Sklera einher. Die Sklera ist ein gefäßarmes, faseriges Gewebe, und die tiefe Skleritis ist eine seltene Erkrankung. Sie kann ein- oder beidseitig auftreten. Die Ursachen werden in idiopathische, mit systemischen Erkrankungen assoziierte, infektiöse und postoperative unterteilt.
Die Inzidenz wird mit 1,6 bis 5,5 pro 100.000 Personenjahre angegeben 5). Frauen sind häufiger betroffen; bei nicht-nekrotisierender diffuser oder nodulärer Skleritis liegt das Durchschnittsalter bei etwa 40 Jahren, bei nekrotisierender Skleritis bei etwa 60 Jahren. Die bilaterale Beteiligung bei nekrotisierender Skleritis beträgt etwa 60 %. Nicht-infektiöse Skleritis macht die Mehrheit der Fälle aus und tritt häufig als Augenmanifestation systemischer entzündlicher Erkrankungen auf. Infektiöse Skleritis ist mit 5–10 % selten, hat jedoch eine schlechte Prognose 7).
Laut einer multizentrischen Studie der japanischen Leitlinie zur Behandlung von Uveitis machte die Skleritis 235 (6,2 %) von 3.810 Fällen in der Uveitis-Ambulanz aus und ist damit nach der akuten anterioren Uveitis (6,6 %) die zweithäufigste Erkrankung 8).
Zur Klassifikation der Skleritis nach klinischen Befunden wird häufig die klassische Watson-Klassifikation (Watson et al., 1976) verwendet. Sie unterscheidet grob zwischen anteriorer und posteriorer Skleritis, wobei die anteriore Skleritis je nach Form weiter in drei Typen unterteilt wird.
Klassifikation
Krankheitstyp
Merkmale
Vordere Skleritis
Diffus
Häufigste Form. Diffuse Rötung durch Erweiterung und Schlängelung der Skleralgefäße
Vordere Skleritis
Nodulär
Dunkelrote Skleraknoten. Bevorzugt im Limbus- und Lidspaltenbereich.
Vordere Skleritis
Nekrotisierend (entzündlich)
Skleranekrose, Ausdünnung, Perforationsrisiko
Vordere Skleritis
Nekrotisierend (nicht entzündlich)
Skleromalazie perforans. Schmerzlos
Hintere Skleritis
—
Selten. Etwa 4 % aller Fälle5)
Die diffuse Skleritis ist am häufigsten, gefolgt von der nodulären Skleritis. Die nekrotisierende Skleritis und die hintere Skleritis sind selten. Bei einem Rückfall tritt meist derselbe Krankheitstyp auf, aber etwa 10% verschlechtern sich. Wenn die zugrunde liegende systemische Erkrankung unbehandelt bleibt, kommt es nicht selten zu wiederholten Rückfällen an derselben Stelle der Sklera. Etwa 10% der nodulären Skleritiden entwickeln sich im Verlauf zu einer nekrotisierenden Skleritis.
Eine Sonderform der nekrotisierenden Skleritis, die kaum Entzündungssymptome aufweist, wird als Skleromalazie (Scleromalacia perforans) bezeichnet. Sie tritt bevorzugt bei Patienten mit langjähriger rheumatoider Arthritis auf und führt ohne Rötung oder Schmerzen zu einer langsamen Ausdünnung der Sklera. Obwohl der englische Name „perforans“ (durchbohrend) enthält, wird die Augenform oft durch eine dünne Fasermembran erhalten.
QWas ist der Unterschied zwischen Episkleritis und Skleritis?
A
Die Episkleritis ist eine Entzündung der oberflächlichen Gefäße wie des Tenon-Kapsel-Gefäßplexus, mit leichter Rötung, ohne Schmerzen und ohne Beeinträchtigung des Sehvermögens. Die Skleritis ist eine Entzündung der tiefen Gefäße, begleitet von starken Augenschmerzen und dunkelroter Rötung. Die Rötung der Episkleritis klingt nach der Gabe von 1:1000 verdünntem Epinephrin-Augentropfen ab, während die Rötung der Skleritis nicht abklingt, was zur Unterscheidung dient.
Smeller L, Toth-Molnar E, Sohar N. Optical Coherence Tomography: Focus on the Pathology of Macula in Scleritis Patients. J Clin Med. 2023;12(14):4825. Figure 1. PMID: 37510941; PMCID: PMC10381547; DOI: 10.3390/jcm12144825. License: CC BY 4.0.
Vorderabschnittsbild mit diffuser Rötung und deutlicher Erweiterung und Schlängelung tiefer Gefäße in der temporalen Sklera des linken Auges. Entspricht den Rötungsmustern und Gefäßbefunden der verschiedenen Krankheitstypen, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt werden.
Starke Augenschmerzen: Charakteristisch sind bohrende Tiefenschmerzen. Die Schmerzen können so stark sein, dass sie den Schlaf stören.
Ausstrahlender Schmerz: Der Schmerz strahlt in Ohr, Gesicht, Kiefer und Schläfe aus. Besonders ausgeprägt bei diffuser Skleritis.
Nächtliche Verschlechterung und Schmerzen bei Augenbewegungen: Die Schmerzen verschlimmern sich nachts und bei Augenbewegungen.
Druckschmerz: Bei der Palpation wird häufig über Druckschmerz geklagt.
Rötung: Es wird eine Rötung mit pochenden starken Schmerzen wahrgenommen.
Sehverschlechterung: Bei schweren Fällen, die bis zur nekrotisierenden Skleritis fortgeschritten sind, oder bei hinterer Skleritis mit Schädigung der Netzhaut oder des Sehnervs wird sie oft erstmals bemerkt.
Besonderheit der perforierenden Skleromalazie: Es treten plötzlich Nekroseherde der Sklera auf, fast ohne Entzündungssymptome, Rötung oder Schmerzen, oder es wird eine Freilegung der Uvea aufgrund eines Skleradefekts bemerkt.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Erweiterung und Schlängelung tiefer Gefäße: Aufgrund der Entzündung der Skleralgefäße erweitern sich der episklerale und der intraklerale Gefäßplexus. Die Skleralgefäße sind nicht verschieblich.
Violett-bläuliche Verfärbung: eine für die Skleritis charakteristische Farbveränderung. Im Gegensatz zum hellen Rot der Konjunktivitis oder Episkleritis zeigt sie eine dunkelrote bis violett-blaue Färbung. Sie ist mit bloßem Auge bei natürlichem Licht besser zu erkennen als mit der Spaltlampe. Bei länger bestehenden Fällen kann die Sklera durch umschriebene oder diffuse Ausdünnung bläulich-schwarz erscheinen.
Epinephrin-Augentropfentest: Bei Verdünnung 1:1000 führt Epinephrin-Augentropfen nicht zum Abklingen der tiefen Gefäßinjektion. Oberflächliche konjunktivale und episklerale Injektion klingt jedoch ab, was zur Unterscheidung von Konjunktivitis und Episkleritis nützlich ist.
Keine Befunde an der Lidbindehaut: Selbst bei schweren Fällen zeigt die Lidbindehaut keine Entzündungszeichen, was die Abgrenzung zur Konjunktivitis erleichtert.
Druckschmerz bei Palpation: Ein wichtiges Hilfsmittel zur Unterscheidung von Konjunktivitis und Episkleritis.
Unterschiede der Befunde nach Krankheitstyp: Die Merkmale variieren je nach Typ und können in der Regel bereits bei der ersten Spaltlampenuntersuchung unterschieden werden.
Diffuse Skleritis
Hyperämie: Eine diffuse, starke Rötung durch Erweiterung und Schlängelung der Skleragefäße. Sie erstreckt sich zirkulär oder ist auf einen Quadranten oder mehr lokalisiert.
Schmerzen: Starke Schmerzen, die in Gesicht und Schläfe ausstrahlen, sind charakteristisch und so stark, dass sie den Schlaf stören.
Besondere Befunde: Keine Knötchen, Erhebungen, Nekrosen oder Ausdünnung. Es können Chemosis, Lidödem, anteriore Uveitis und erhöhter Augeninnendruck auftreten.
Nodulöse Skleritis
Knoten (Noduli): Einzelne oder multiple dunkelrote Knoten. Bevorzugt treten sie im Lidspaltenbereich in der Nähe des Limbus auf.
Palpation: Der Knoten ist nicht verschieblich und druckempfindlich.
Anamnese: Viele Patienten haben eine Vorgeschichte mit Herpes Zoster ophthalmicus. Etwa 10% entwickeln eine nekrotisierende Skleritis, die bei frühzeitiger Behandlung unter Hinterlassung kleiner Narbenherde ausheilt.
Nekrotisierende Skleritis
Skleranekrose: Anfangs umschriebene weiße bis gelbe avaskuläre Areale (Skleranekroseherde). Begleitet von starker Erweiterung und Schlängelung der Skleragefäße sowie Einschmelzung.
Ausdünnung: Die Aderhaut wird so dünn, dass sie durchscheinend wird, und bei weiterem Fortschreiten kann es zu einer Perforation des Augapfels kommen. Die ausgedünnte Stelle bleibt auch nach Abklingen der Entzündung bestehen.
Prognose: Das Erkrankungsalter liegt in den 60er Jahren und ist hoch, etwa 60% sind beidseitig betroffen. Ohne frühzeitige Behandlung drohen Erblindung und Schwierigkeiten, den Augapfel zu erhalten.
Hintere Skleritis
Epidemiologie: Das durchschnittliche Erkrankungsalter liegt bei etwa 50 Jahren, Frauen sind etwa doppelt so häufig betroffen wie Männer. In 30–40 % der Fälle tritt die Erkrankung beidseitig auf.
Ausstrahlende Symptome: Bei einer Begleitmyositis der äußeren Augenmuskeln treten Doppelbilder, Schmerzen bei Augenbewegungen, Exophthalmus und Ptosis auf.
Wenn die Entzündung von der Skleritis auf die Hornhaut übergreift, können periphere Hornhautinfiltrate und Ulzerationen auftreten. Auch eine vordere Uveitis kann als Komplikation hinzukommen. Da die Skleritis fast ausnahmslos auch die Episklera betrifft, sind die Befunde einer Episkleritis ebenfalls vorhanden.
Die hintere Skleritis wird oft spät diagnostiziert, da die Läsion tief im Augenhintergrund liegt. Sie kann gleichzeitig im vorderen und hinteren Bereich auftreten oder zeitlich versetzt. Etwa ein Drittel der Fälle von hinterer Skleritis ist mit einer vorderen Skleritis assoziiert, und im Verlauf der hinteren Skleritis wird bei etwa 70 % eine vordere Skleritis beobachtet 1). Bei Fällen mit begleitender vorderer Skleritis besteht ein starker Zusammenhang mit systemischen Erkrankungen.
Aderhautfalten: Es zeigen sich Falten in der Aderhaut1).
Papillenödem: Tritt auf, wenn sich die Entzündung auf das Orbitagewebe oder den Sehnerv ausbreitet, und erfordert eine sofortige Behandlung, um dauerhafte Sehstörungen zu vermeiden.
T-Zeichen im B-Mode-Ultraschall: Aufgrund der Skleraverdickung und der Flüssigkeitsansammlung unter der Tenon-Kapsel wird die Grenze zwischen Sehnerv und Sklera eckig dargestellt1). Dies ist der spezifischste Ultraschallbefund bei hinterer Skleritis.
Verwechslung mit Aderhauttumor: Eine hintere Skleritis kann als Aderhauttumor vorgestellt werden und ist eine Ursache für ein Pseudomelanom (falsches Melanom) 1).
Begleitende externe Ophthalmomyositis: Wenn die Entzündung auf die äußeren Augenmuskeln übergreift, führt dies zu Doppelbildern, Schmerzen bei Augenbewegungen und Rötung um die Ansatzstellen der äußeren Augenmuskeln.
QWarum wird die hintere Skleritis leicht übersehen?
A
Bei der hinteren Skleritis sind die vorderen Augenabschnitte unauffällig, und es gibt Fälle, in denen Patienten nur wegen Augenschmerzen, Kopfschmerzen und Sehverschlechterung einen Arzt aufsuchen. Fundusbefunde wie Aderhautfalten und exsudative Netzhautablösung werden oft erst durch das T-Zeichen im B-Mode-Ultraschall oder die Beurteilung der Aderhautdicke mittels OCT sicher diagnostiziert 1). Es kann auch mit einem Aderhauttumor verwechselt werden, daher ist eine umfassende Differenzialdiagnose wichtig.
Bei bis zu 50 % der Skleritis-Fälle liegt eine systemische Autoimmunerkrankung vor. Bleibt die zugrunde liegende systemische Erkrankung unbehandelt, kommt es nicht selten zu wiederholten Schüben an derselben Stelle der Sklera. Die Ursachen der nekrotisierenden Skleritis sind häufig rheumatische Erkrankungen, Vaskulitis und Bluterkrankungen.
Kollagenosen und rheumatische Erkrankungen
Rheumatoide Arthritis (RA): Die am häufigsten assoziierte systemische Erkrankung. Sie kann zu nekrotisierender Skleritis oder perforierender Skleromalazie führen. Typisch ist sie bei Patienten unter Langzeittherapie.
Rezidivierende Polychondritis: Sie verursacht sowohl anteriore als auch posteriore Skleritis und verläuft mit Remissionen und Exazerbationen.
Vaskulitis und Sonstiges
Granulomatose mit Polyangiitis (GPA): Sie geht mit nekrotisierender Skleritis und perforierender Skleromalazie einher und hat einen schweren Verlauf. Als ANCA-assoziierte Vaskulitis können Augenveränderungen das erste Symptom sein3).
Takayasu-Arteriitis: Selten kommt es zu einer Skleritis, die zur Diagnose einer systemischen Vaskulitis führen kann4).
Polyarteritis nodosa (PAN): Kann mit nekrotisierender Skleritis einhergehen.
Darüber hinaus wurde über Assoziationen mit Sarkoidose, Morbus Behçet, Morbus Crohn, Colitis ulcerosa, Psoriasis-Arthritis, Sklerodermie, Dermatomyositis, SAPHO-Syndrom, Schilddrüsenerkrankungen, Aortitis-Syndrom, interstitieller Nephritis, Vogt-Koyanagi-Harada-Krankheit und Multipler Sklerose berichtet. Bei nodulärer Skleritis haben viele Patienten eine Vorgeschichte von Herpes Zoster ophthalmicus. Bei posteriorer Skleritis kann sie selten als Augenmanifestation eines systemischen Lymphoms oder multiplen Myeloms auftreten, was besondere Aufmerksamkeit erfordert.
Infektiöse Skleritis macht nur 5–10 % aller Fälle aus, hat jedoch eine äußerst schlechte Prognose7). Berichten zufolge verlieren etwa 50 % der Patienten mit infektiöser Skleritis das funktionelle Sehvermögen, und bei etwa 27 % kommt es zur Enukleation oder Eviszeration des Auges7).
Pseudomonas aeruginosa: Der häufigste Erreger in Europa und Nordamerika 7). Die Skleranekrose schreitet rasch voran und zeigt das Bild einer eitrigen Skleromalazie.
Nocardia spp.: Tritt nach Verletzungen oder bei immungeschwächten Patienten auf 2). Charakteristisch ist ein chronischer Verlauf mit wiederholten Rückfällen und Remissionen; selbst wenn die Knoten abklingen, verbleiben Bakterien in der Tiefe 2).
Moraxella-Arten: Seltene Erreger, die bei Immunschwäche als opportunistische Infektion auftreten7).
Sonstige: Auch Infektionen mit Pilzen, Tuberkulose, Syphilis, Herpesviren u. a. wurden berichtet. In Regionen mit hoher Tuberkulose-Inzidenz wird empfohlen, vor einer systemischen Steroidtherapie mittels Tuberkulin-Hauttest eine Tuberkulose auszuschließen.
Die meisten Fälle von infektiöser Skleritis werden durch freiliegende Nähte oder Sklera-Buckel-Material nach Augenoperationen verursacht und treten einseitig auf.
Augenoperationen können eine nekrotisierende Skleritis auslösen. Typische Auslöser sind Pterygium-Operationen, Kataraktoperationen, Skleraeindellungen, Schieloperationen und Trabekulektomien. Besonders häufig tritt sie nach einer Pterygium-Exzision mit Mitomycin C auf. Der Zeitraum bis zum Auftreten reicht von wenigen Tagen bis zu mehreren Jahren nach der Operation, und Fälle mit Auftreten mehrere Jahre postoperativ sind nicht selten.
Die Antimetaboliten Mitomycin C (MMC) und 5-Fluorouracil (5-FU) wurden zur Verhinderung des Rezidivs nach Pterygium-Operation und zur Narbenbildung von Glaukomfiltrationsblasen eingesetzt. MMC-Augentropfen können Monate bis Jahre nach der Operation zu Skleraverkalkung oder perforierender Skleromalazie führen, weshalb ihre Verwendung als Augentropfen in den 1980er Jahren eingestellt wurde. Bei aktuellen Glaukom- und Pterygium-Operationen wird hauptsächlich eine einmalige intraoperative Applikation von niedrig konzentriertem MMC (0,02–0,04 %) verwendet. Postoperativ kann es jedoch zu einer Blässe der Sklera, Gefäßverengung und avaskulären Zonen im Operationsbereich kommen, die eine Grundlage für zukünftige Skleromalazie bilden können.
QWelche systemischen Erkrankungen sind damit verbunden?
A
Rheumatoide Arthritis tritt am häufigsten auf, daneben kommen Autoimmunerkrankungen wie Granulomatose mit Polyangiitis (GPA), systemischer Lupus erythematodes, Polyarteriitis nodosa, rezidivierende Polychondritis, Takayasu-Arteriitis und Sarkoidose vor. Bei bis zu 50 % der Patienten mit Skleritis wird eine systemische Erkrankung festgestellt. Bei nekrotisierender Skleritis ist die Rate an rheumatischen Erkrankungen, Vaskulitiden und Bluterkrankungen noch höher.
Makroskopische Beobachtung bei natürlichem Licht: Im Gegensatz zur hellroten Rötung bei Konjunktivitis oder Episkleritis zeigt die Skleritis eine dunkelrote bis bläulich-violette Färbung. Bei länger bestehenden Fällen erscheint die Sklera durch Ausdünnung bläulich-schwarz. Diese Farbveränderungen sind mit bloßem Auge im hellen Raum besser zu erkennen als mit der Spaltlampe.
Spaltlampenuntersuchung: Beurteilung von Erweiterung und Schlängelung der Skleragefäße, Vorhandensein von Knoten, dunkelrote Befunde der Skleraknoten, Ausdünnung, Nekrose und Perforation. Dass an der Lidbindehaut keine Entzündungszeichen zu finden sind, ist ein Unterscheidungsmerkmal zur Konjunktivitis.
Epinephrin-Augentropfentest: Bei der Anwendung von 1:1000 verdünntem Epinephrin verschwindet die tiefe Sklerahyperämie nicht. Dies ist wichtig für die Unterscheidung von Episkleritis und konjunktivaler Hyperämie.
Palpation: Mit einem Wattestäbchen oder ähnlichem wird die Bindehaut berührt, um Druckempfindlichkeit festzustellen. Dies hilft bei der Unterscheidung von Konjunktivitis und Episkleritis.
B-Mode-Ultraschalluntersuchung: Sie ist für die Diagnose der hinteren Skleritis unerlässlich. Charakteristisch sind Skleraverdickung, Skleraknoten und das T-Zeichen durch Flüssigkeitsansammlung unter der Tenon-Kapsel1). Sie ist auch nützlich zur Abgrenzung von Aderhauttumoren.
Fluorescein-Fluoreszenz-Sklera-Angiographie: Anhand des Vorhandenseins oder Fehlens von skleralen Nichtperfusionsarealen kann eine nekrotisierende Skleritis unterschieden werden.
CT/MRT: Wird zur Beurteilung der Skleraverdickung bei posteriorer Skleritis und der extraokularen Myositis sowie zur Abgrenzung von intrakraniellen Läsionen eingesetzt.
Bei nekrotisierender Skleritis oder therapierefraktären Fällen ist die Untersuchung auf ANCA-assoziierte Vaskulitis besonders wichtig3). In Regionen mit hoher Tuberkuloseprävalenz sollte bei Skleritis, die auf eine lokale Steroidtherapie nicht anspricht, vor einer systemischen Behandlung ein Tuberkulin-Hauttest durchgeführt werden.
Episkleritis: Es handelt sich um eine Entzündung der oberflächlichen Blutgefäße, die Rötung ist mild, schmerzlos und klingt nach Phenylephrin-Augentropfen ab.
Bindehautentzündung: Die Rötung ist am stärksten im Bereich des Bindehautfornix und nimmt zur Hornhaut hin ab. Sie geht mit Augenausfluss und Auffälligkeiten der Lidbindehaut einher.
MALT-Lymphom: Ein lachsfarbener Tumor, der bevorzugt im Fornix conjunctivae auftritt. Da der Tumor subkonjunktival liegt, sind die Skleralgefäße nicht sichtbar, was zur Abgrenzung von einer Skleritis dient.
Hornhauterkrankungen: Es ist eine Abgrenzung von peripheren Hornhautinfiltraten, die von einer Skleritis ausgehen, von Mooren-Ulkus und staphylokokkenbedingten peripheren Hornhautinfiltraten erforderlich.
Tenon-Kapsel-Entzündung (Tenonitis): Wird als eine Form der Episkleritis betrachtet, und die Unterscheidung zwischen beiden ist schwierig.
Orbitaspitzensyndrom: Bei einem Karotis-Sinus-cavernosus-Fistel kommt es zu einer Stauung und Erweiterung der konjunktivalen und skleralen Venen, begleitet von pulsierendem Exophthalmus und Doppelbildern.
Vogt-Koyanagi-Harada-Krankheit: Eine Erkrankung, die schwer von der hinteren Skleritis zu unterscheiden ist. Sie zeigt sich beidseitig mit granulomatöser anteriorer Uveitis und einer Aderhautverdickung im OCT.
Aderhauttumor: Die knotigen Läsionen der hinteren Skleritis können als Aderhauttumoren vorgestellt werden 1) und werden mittels Ultraschall, MRT und OCT differenziert.
Die Behandlung der Skleritis basiert hauptsächlich auf Steroiden, wobei je nach Form und Schweregrad lokale Behandlung, systemische Therapie, Immunsuppressiva, Biologika und chirurgische Eingriffe schrittweise kombiniert werden. Bei Patienten mit systemischen Erkrankungen ist die Zusammenarbeit mit Rheumatologen und Spezialisten für Kollagenosen unerlässlich.
Orale NSAR (erste Wahl)
Indikation: Initialtherapie bei leichter bis mittelschwerer diffuser oder nodulärer Skleritis.
Verschreibungsbeispiel: Celecoxib (COX-2-Hemmer) 100 mg 2-mal täglich oral oder Indomethacin 50 mg 3-mal täglich. Wirkt oft sehr gut gegen Schmerzen und ist auch zur Kontrolle von Entzündungen wirksam.
Achtung: Achten Sie auf gastrointestinale Blutungen, Nierenfunktionsstörungen und Asthmaanfälle. Wenn keine Kontraindikationen wie Asthma vorliegen, von Anfang an aktiv kombinieren.
Lokale Steroidtherapie
Augentropfen: 0,1% Betamethason-Natriumphosphat-Augentropfen 4- bis 6-mal täglich anwenden. Je nach Fall wird zusätzlich vor dem Schlafengehen Betamethason-Fradiomycin-Salbe in den Bindehautsack gegeben.
Subkonjunktivale Injektion: Triamcinolonacetonid 40 mg/ml einmalig 0,1 ml (maximal einmal monatlich) oder Dexamethason 3,3 mg/ml einmalig 0,3 ml alle 1–2 Wochen für mehrere Male.
Achtung: Bei nekrotisierender Skleritis sollte die Injektion unter Vermeidung dünner Stellen erfolgen.
Systemische Steroidtherapie
Orale Einnahme: Prednisolon 0,5–1 mg/kg/Tag (bei leichten Fällen, die nicht auf NSAR ansprechen, 20–30 mg in 2 geteilten Dosen mit ausschleichender Dosierung; bei schwerer nodulärer, nekrotisierender oder posteriorer Skleritis 30–60 mg/Tag mit ausschleichender Dosierung).
Pulstherapie: 1.000 mg Methylprednisolon pro Tag als intravenöse Infusion über 3 Tage, gefolgt von einer ausschleichenden Therapie. Indiziert bei nekrotisierender Skleritis und schweren Fällen.
Hinweis: Die Ausschleichung erfolgt in der Regel über 1–2 Wochen, bei schweren Fällen über 2–3 Monate.
Immunsuppressiva und Biologika
Cyclosporin: Beginn mit 5 mg/kg/Tag in 2 Dosen, Einstellung des Blutspiegeltals auf etwa 100–150 ng/mL. Achtung auf Nierenfunktionsstörungen, regelmäßige Blutuntersuchungen erforderlich.
Auswahl bei systemischen Erkrankungen: Bei rheumatoider Arthritis wird häufig Methotrexat eingesetzt, bei systemischem Lupus erythematodes oder systemischer Vaskulitis Cyclophosphamid. Azathioprin ist bei Skleritis weniger wirksam.
Biologika: Es gibt Berichte über die Anwendung von Infliximab (Anti-TNF-α-Antikörper, Remicade®) und Rituximab (Anti-CD20-Antikörper, Rituxan®) bei therapierefraktären Fällen3).
Die Behandlung beginnt mit oralen NSAIDs und 0,1% Betamethason-Augentropfen. Bei unzureichender Wirkung werden immunsuppressive Augentropfen hinzugefügt, und falls weiterhin unzureichend, erfolgt eine subkonjunktivale Injektion von 0,3 ml Dexamethason oder 0,1–0,2 ml Triamcinolonacetonid unter Vermeidung dünner Sklerabereiche. Bei schlechtem Ansprechen auf die lokale Behandlung wird Prednisolon 20–30 mg/Tag für 1–2 Wochen in ausschleichender Dosierung fortgesetzt.
Da es innerhalb kurzer Zeit zu einer Skleraverdünnung und Perforation kommen kann, wird bereits in der Initialtherapie Prednisolon oral in einer Dosis von 0,5–1 mg/kg/Tag begonnen. Bei Patienten, die unzureichend auf orale Steroide ansprechen oder wiederholt Rezidive erleiden, wird in Zusammenarbeit mit der Rheumatologie das für die begleitende systemische Erkrankung optimale Immunsuppressivum ausgewählt. Bei gleichzeitiger Gabe von Ciclosporin 5 mg/kg/Tag in zwei Einzeldosen wird der Blutspiegel-Talspiegel auf 100–150 ng/mL eingestellt. Bei Skleritis im Rahmen eines Morbus Behçet mit neurologischer Beteiligung ist Ciclosporin aufgrund der Gefahr einer Verschlechterung der neurologischen Symptome kontraindiziert. In schweren Fällen wird eine Pulstherapie mit Methylprednisolon 1.000 mg/Tag als intravenöse Infusion über 3 Tage durchgeführt, nachdem infektiöse Ursachen ausreichend ausgeschlossen wurden.
Bei einer gegen Immunsuppressiva resistenten Skleritis sollte der Einsatz von Biologika in Betracht gezogen werden. TNF-α-Inhibitoren haben sich bei skleraler Uveitis als wirksam erwiesen, sind jedoch nicht bei allen Skleritisformen wirksam; unter Etanercept wurden Fälle von paradoxen Reaktionen berichtet, die Augenentzündungen einschließlich Skleritis auslösen oder verschlimmern. Vor und nach der Anwendung von Immunsuppressiva und Biologika sind eine umfassende systemische Untersuchung und die Zusammenarbeit mit der Inneren Medizin unerlässlich.
Die systemische Verabreichung von Kortikosteroiden ist die Hauptbehandlung. Prednisolon wird von 30–50 mg/Tag beginnend schrittweise reduziert und zur Schmerzkontrolle auf 2–3 Dosen pro Tag aufgeteilt eingenommen. Bei begleitender Entzündung des vorderen Augenabschnitts werden zusätzlich kortikosteroidhaltige Augentropfen eingesetzt. Wenn durch orale Einnahme keine Beruhigung der Entzündung erreicht wird, wird nach ausreichendem Ausschluss infektiöser Ursachen eine Steroid-Pulstherapie durchgeführt. Bei Resistenz gegen die Pulstherapie oder bei einem Rückfall während der Dosisreduktion wird die aktive Gabe von Immunsuppressiva in Betracht gezogen. Als Behandlungsbeispiele werden Azathioprin 1–2 mg/kg oder Methotrexat (Rheumatrex®) 6 mg/Woche gewählt. Bei nekrotisierender Skleritis kann eine Cyclophosphamid-Pulstherapie erforderlich sein, und die Zusammenarbeit mit einem Internisten ist besonders wichtig.
Die subtenonale Triamcinolonacetonid-Injektion (STTA) wird auch bei posteriorer Skleritis eingesetzt, birgt jedoch selten das Risiko von Durchblutungsstörungen des Sehnervs und der Netzhaut-Aderhaut6). Bei älteren Patienten mit Gefäßbrüchigkeit und bei Glaukomaugen ist eine sorgfältige Indikationsstellung erforderlich 6).
Bei infektiöser Skleritis basiert die Behandlung auf der Identifizierung des Erregers und einer darauf abgestimmten Antibiotikatherapie 2)7).
Selektive Therapie nach Erregeridentifikation: Bei Nocardia-Infektion werden verstärkte Amikacin-Augentropfen und orales Sulfamethoxazol-Trimethoprim langfristig kombiniert. Chirurgisches Débridement kann wiederholt erforderlich sein2). Bei Pseudomonas aeruginosa-Infektion schreitet die Skleranekrose (eitrige Skleromalazie) rasch voran, daher werden eine intensive Therapie mit Aminoglykosiden oder Chinolonen sowie eine sofortige chirurgische Behandlung durchgeführt.
Entfernung von exponierten Nähten und Plombenmaterial: Exponierte Nähte, die die Infektionsquelle darstellen, sollten sofort entfernt werden. Auch Skleraplombenmaterial sollte, wenn es nicht auf die medikamentöse Behandlung anspricht, innerhalb von ein bis zwei Wochen entfernt werden, um einer Endophthalmitis vorzubeugen.
Entscheidung über den Einsatz von Steroiden: Da Steroide das Risiko einer Infektionsverschlechterung bergen, werden sie erst nach ausreichendem Ausschluss einer infektiösen Ursache eingesetzt. Wenn die Antibiotika ansprechen, aber die Entzündung anhält, können Steroide unter Berücksichtigung der Leukozytenzahl und des CRP-Verlaufs eingesetzt werden.
Fungale infektiöse Skleritis: Die Behandlung erfolgt analog zur medikamentösen Therapie der Pilzkeratitis.
Eine nekrotisierende Skleritis oder eine perforierende Skleromalazie sowie eine infektiöse Skleritis, die nicht auf eine medikamentöse Behandlung anspricht, sind Indikationen für eine chirurgische Therapie. Wenn die Skleranekrose oder -erweichung ein bestimmtes Ausmaß überschreitet, wird die Wiederherstellung der Bulbusform und der Sehfunktion schwierig, daher ist eine frühzeitige Operation bei noch kleiner Nekrosefläche wünschenswert.
Die wichtigsten Punkte der Operation sind die folgenden drei.
Vollständige Resektion des Skleranekroseherds einschließlich des angrenzenden gesunden Gewebes
Reparatur und Auffüllung der Läsion durch konservierte Skleratransplantation: Konservierte Sklera eignet sich aufgrund ihrer Festigkeit und der Fähigkeit, die Augenwandform zu erhalten, gut als Füllmaterial. Konservierte Hornhaut schmilzt häufig.
Vollständige Abdeckung des transplantierten Sklerastücks durch die Bindehaut
Bei ausgedehnter Bindehautnekrose oder peripherem Hornhautulkus wird eine autologe Bindehauttransplantation oder eine Transplantation von Hornhautepithel vom anderen Auge durchgeführt. Zur postoperativen Behandlung werden orales Ciclosporin und 1% Sandimmun® Augentropfen (Krankenhauszubereitung) eingesetzt, um das Einwachsen des Transplantats zu fördern und ein Wiederauftreten der Skleritis zu verhindern. Auch bei nach MMC- oder 5-FU-Anwendung aufgetretener Skleromalazie sollte, solange keine gesicherte Wirksamkeit von immunsuppressiven Augentropfen oder Biologika vorliegt, frühzeitig eine konservative Skleratransplantation durchgeführt werden, solange die Erweichungsstelle noch klein ist.
Systemisches Management und Überwachung von Nebenwirkungen
Bei einer Langzeittherapie mit Steroiden und Immunsuppressiva müssen Augeninnendruckerhöhung, Leber- und Nierenfunktionsstörungen, Blutzuckeranstieg und der Blutspiegel von Cyclosporin regelmäßig überwacht werden. Bei erhöhtem Blutzucker kann eine systemische Behandlung in Zusammenarbeit mit einem Internisten erforderlich sein, bei der Steroide in Kombination mit Insulin eingesetzt werden. Bei Augenschmerzen werden entzündungshemmende Schmerzmittel verabreicht. Da eine Skleritis durch eine Infektion oder eine infektionsallergische Reaktion ausgelöst werden kann, werden bei der Erstbehandlung auch antibiotische Augentropfen und orale Antibiotika eingesetzt.
QWie wird eine infektiöse Skleritis behandelt?
A
Die infektiöse Skleritis hat eine schlechte Prognose, und eine frühzeitige Gabe von Antibiotika basierend auf der Identifizierung des Erregers und seiner Empfindlichkeit ist unerlässlich. Bei einer Pseudomonas-Infektion schreitet die Erkrankung schnell voran, daher sind eine intensive Antibiotikatherapie und ein chirurgisches Débridement erforderlich 7). Wenn freiliegende Fäden oder Buckelmaterial die Ursache sind, sollten diese schnell entfernt werden. Steroide sollten bei möglicher Infektion vorsichtig eingesetzt werden, da sie das Risiko einer Verschlimmerung der Infektion bergen.
6. Pathophysiologie und detaillierter Pathomechanismus
Die Sklera ist ein gefäßarmes faseriges Gewebe, und die Inzidenz einer tiefen Skleritis ist gering. Da die Sklera jedoch innerviert ist, verursacht eine Entzündung starke Augenschmerzen. Die Sklera ist an der Ansatzstelle der geraden Augenmuskeln mit etwa 0,3 mm am dünnsten und stellt eine Prädilektionsstelle für Nekrose und Perforation dar. Da die Sklera keine Barriere aufweist, können subkonjunktival oder unter der Tenon-Kapsel injizierte Medikamente durch Diffusion in das Auge gelangen, weshalb subkonjunktivale Steroidinjektionen eine therapeutische Option darstellen.
Die Pathologie der mit Autoimmunerkrankungen assoziierten Skleritis ist durch eine zonale granulomatöse Nekrose gekennzeichnet. Im Zentrum des Granuloms findet sich fibrinoides Material, umgeben von Epitheloidzellen und mehrkernigen Riesenzellen.
Bei Skleritis ist die Infiltration von Entzündungszellen, einschließlich T-Zellen und Makrophagen, erhöht. T-Zellen und Makrophagen infiltrieren das tiefe episklerale Gewebe, und um die Blutgefäße herum bilden sich B-Zell-Cluster. Die erhöhte Expression von HLA-DR und IL-2-Rezeptor auf T-Zellen deutet auf eine Beteiligung der zellvermittelten Immunantwort hin.
Plasmazellen sind an der Produktion von Matrix-Metalloproteinasen (MMPs) und TNF-α beteiligt. Bei der nekrotisierenden Skleritis findet sich eine Vaskulitis mit fibrinoider Nekrose, die eine neutrophile Infiltration der Gefäßwand zeigt. Der Entstehungsmechanismus der endogenen Skleritis deutet auf eine Beteiligung des Immunsystems hin, insbesondere auf zellvermittelte Immunreaktionen.
Nicht-nekrotisierende Skleritis (diffus, nodulär): Vaskulitis ist nicht ausgeprägt, nicht-granulomatöse Entzündung steht im Vordergrund. Bei der nodulären Form zeigen sich zentrale Fibrinoidnekrose und periphere Anordnung von Epitheloidzellen.
Nekrotisierende Skleritis: Es zeigen sich kleine Nekroseherde und eine vorwiegend lymphozytäre, plasmazelluläre und makrophagenvermittelte nicht-granulomatöse Entzündung. Charakteristisch sind Vaskulitis mit Fibrinoidnekrose und neutrophile Infiltration.
Infektiöse Skleritis: Neben der nekrotisierenden Entzündung bilden sich Mikroabszesse. Bei einer Nocardia-Infektion verbleiben die Bakterien auch nach Abklingen der Knoten tief im Gewebe, was zu wiederholten Rückfällen führt2).
Skleromalazie perforans (Scleromalacia perforans): Tritt bei Patienten mit langjähriger rheumatoider Arthritis oder verwandten Erkrankungen auf. In der Nähe des Limbus zeigen sich nekrotische Skleraplaques ohne Stauung, die zu einer langsamen Ausdünnung der Sklera und Freilegung der Uvea führen.
Nach der COVID-19-Impfung oder -Infektion wurde eine Serie von 8 Fällen von posteriorer Skleritis berichtet, die fälschlicherweise als Aderhautmelanom diagnostiziert und überwiesen wurden 5). Der durchschnittliche Zeitraum von der letzten Impfung bis zum Auftreten betrug 132 Tage, von der COVID-19-Infektion bis zum Auftreten 14 Tage 5). Die meisten Fälle klangen innerhalb von 2 Monaten spontan ab, mit geringen Auswirkungen auf das Sehvermögen 5).
Rituximab bei therapierefraktärer ANCA-assoziierter Skleritis
Für eine ANCA-assoziierte nekrotisierende Skleritis, die resistent gegen konventionelle immunsuppressive Therapie (Steroide + Cyclophosphamid) ist, wurden Fälle berichtet, in denen Rituximab (Anti-CD20-Antikörper) sowohl zur Remissionsinduktion als auch zur Erhaltung wirksam war 3). Langzeit-Follow-up-Studien, die die Wirksamkeit von Rituximab bei Augenmanifestationen der Granulomatose mit Polyangiitis im Rahmen einer ANCA-assoziierten Vaskulitis belegen, häufen sich ebenfalls 3).
Es gibt Berichte über Skleritis als Augenmanifestation der Takayasu-Arteriitis, die als diagnostischer Hinweis auf das systemische Aortenentzündungssyndrom beachtet werden muss 4). Bei nekrotisierender Skleritis junger Frauen ist der Ausschluss einer Takayasu-Arteriitis wichtig.
QGibt es einen Zusammenhang mit COVID-19?
A
Nach einer COVID-19-Impfung oder -Infektion wurde eine Fallserie von posteriorer Skleritis berichtet5). Ein kausaler Zusammenhang ist jedoch nicht nachgewiesen, und die meisten Fälle verlaufen selbstlimitierend. Die COVID-19-assoziierte posteriore Skleritis kann mit einem Aderhauttumor verwechselt werden, daher ist die Kenntnis als Differenzialdiagnose wichtig5).
Babu N, Kumar K, Upadhayay A, Kohli P. Nodular posterior scleritis - The great masquerader. Taiwan journal of ophthalmology. 2021;11(4):408-412. doi:10.4103/tjo.tjo_20_21. PMID:35070674; PMCID:PMC8757530.
Chauhan K, Murthy SI, Mitra S. Demystifying nocardial scleritis. BMJ case reports. 2023;16(11). doi:10.1136/bcr-2023-255730. PMID:38011958; PMCID:PMC10685915.
Tahavvori M, Fekri S, Hassanpour K, Sadoughi MM, Javadi M. Isolated ANCA-associated scleritis successfully treated with systemic rituximab; a case report and review of literature. BMC ophthalmology. 2025;25(1):176. doi:10.1186/s12886-025-04027-6. PMID:40197146; PMCID:PMC11974155.
Chittipolu S, Kennard JL, Tumma RS, Doyle AR. Scleritis in Takayasu Arteritis. Cureus. 2023;15(4):e37724. doi:10.7759/cureus.37724. PMID:37206528; PMCID:PMC10191460.
Negretti GS, Zeiger JS, Cherkas E, Shields CL. Posterior scleritis following COVID-19 vaccination or infection simulating uveal melanoma in 8 consecutive patients. Eye (London, England). 2024;38(1):185-191. doi:10.1038/s41433-023-02656-z. PMID:37422535; PMCID:PMC10764359.
Akada M, Muraoka Y, Morooka S, Ishihara K, Hata M, Tsujikawa A. SEVERE CIRCULATORY DISTURBANCE IN OPTIC DISK, RETINA, AND CHOROID AFTER SUB-TENON TRIAMCINOLONE ACETONIDE INJECTION FOR POSTERIOR SCLERITIS. Retinal cases & brief reports. 2025;19(6):789-792. doi:10.1097/ICB.0000000000001642. PMID:39058980; PMCID:PMC12570599.
Dallinga M, Murtagh P, Powell S, Murphy CC. Moraxella nonliquefaciens-associated infectious scleritis. BMJ case reports. 2023;16(5). doi:10.1136/bcr-2022-254113. PMID:37221000; PMCID:PMC10230883.