시신경염

시신경척수염 스펙트럼 장애 (NMOSD)
한눈에 보는 포인트
섹션 제목: “한눈에 보는 포인트”1. 시신경척수염 스펙트럼 장애(NMOSD)란?
섹션 제목: “1. 시신경척수염 스펙트럼 장애(NMOSD)란?”시신경척수염 스펙트럼 장애(NMOSD)는 중추신경계를 침범하는 염증성, 항체 매개성, 자가면역 질환입니다. 이전에는 데빅병(Devic’s disease)이라고도 불렸으며, 오랫동안 다발성 경화증(MS)의 아형으로 간주되었습니다. 그러나 2004년 아쿠아포린4(AQP4)에 대한 자가항체(AQP4-IgG)가 발견되면서 독립적인 질환 단위로 확립되었습니다.
역사적 배경: 1870년 Sir Thomas Clifford Allbutt가 척수염과 시신경 장애의 연관성을 처음 기술했습니다. 이후 연구를 통해 MS와 다른 질환으로 인식되었으며, 현재는 NMOSD라는 포괄적 개념으로 이해되고 있습니다.
역학은 다음과 같습니다:
- 발생률: AQP4 양성 NMOSD의 추정 연간 발생률은 백만 명당 0.4~7.3명입니다1)
- 성비: 남녀 비율은 약 1:9로 여성에서 현저히 높습니다.
- 발병 연령: 주로 중년(40
60세)에 호발하며, 30대 후반40대 초반에 정점을 보입니다. - 인종: 아프리카계 및 아시아계에서 더 흔합니다1)
- 임신과의 연관성: 여성의 약 20~47%가 임신 중 또는 출산/유산 후 1년 이내에 첫 발병을 경험합니다.
Q
NMOSD와 다발성 경화증(MS)은 어떻게 다른가요?
2. 주요 증상 및 임상 소견
섹션 제목: “2. 주요 증상 및 임상 소견”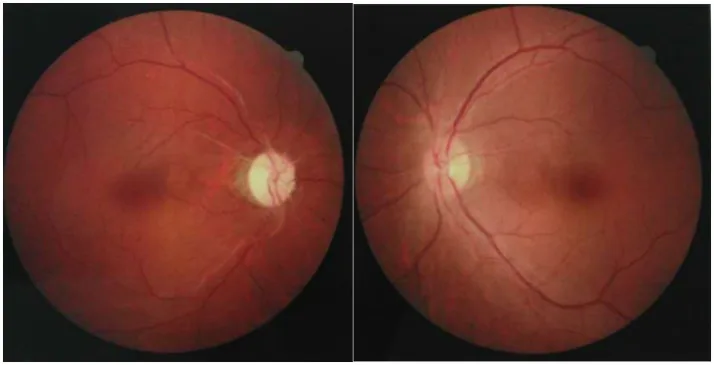
Chuai Y, et al. Paraneoplastic neuromyelitis optica spectrum disorder with dual AQP4-IgG and CRMP5 antibodies following thymectomy: a case report. Front Neurosci. 2026. Figure 1. PMCID: PMC13006573. License: CC BY.
양안의 안저 사진으로, 한쪽 눈에는 시신경 유두의 창백화로 인한 시신경 위축, 반대쪽 눈에는 경미한 시신경 유두 종창이 보입니다. NMOSD에서 나타나는 시신경염의 급성기에서 만성기까지의 시신경 소견이 나타납니다.
자각 증상
섹션 제목: “자각 증상”NMOSD의 증상은 침범된 부위에 따라 다양합니다.
- 급격한 시력 저하: 주요 증상 중 하나입니다. 스테로이드 치료에 저항성이 있는 것이 특징적입니다.
- 안통: 시신경염에 동반되어 약 절반의 증례에서 나타납니다.
- 색각 이상: 적색 채도 감소가 전형적입니다.
- 시야 장애: 중심 암점에 국한되지 않고, 수평 반맹, 양측 이측 반맹, 동측 반맹을 초래하기도 합니다. 시교차와 시삭까지 병변이 미치기 때문입니다.
- 감각 장애 및 하반신 마비: 척수염으로 인한 운동 및 감각 장애입니다.
- 방광직장장애: 척수염에 동반된 자율신경장애
- 난치성 딸꾹질, 메스꺼움 및 구토: 마지막구역(area postrema) 병변에 의한 특징적 증상
- 안구운동장애: 뇌간 병변에 의한 것
- 과다수면증(기면증 유사): 간뇌/시상하부 병변에 의한 것
발병 초기에 인플루엔자 유사 증상(발열, 근육통, 두통)이 나타날 수 있습니다.
임상 소견
섹션 제목: “임상 소견”척수염
LETM(길이방향 광범위 횡단성 척수염): 3개 이상의 척추체 분절에 걸친 연속 병변. AQP4+ NMOSD 환자의 약 85%가 급성 척수염 시 나타냅니다.
완전 척수 증후군: 운동, 감각, 자율신경의 세 경로 모두를 침범합니다.
심각한 기능 장애: 30% 이상의 환자가 발작 최저점에서 휠체어 의존 상태가 됩니다. AQP4+ NMOSD 환자의 37~44%는 결국 보행 보조기가 필요합니다.
마지막구역 증후군
난치성 딸꾹질: 수일에서 수주간 지속되며, 일반적인 항구토제에 반응하지 않습니다.
메스꺼움 및 구토: 마지막구역은 혈액뇌장벽이 없어 AQP4-IgG가 직접 도달하기 쉬운 부위이기 때문입니다.
NMOSD의 핵심 진단 소견: 설명되지 않는 난치성 딸꾹질은 NMOSD를 적극적으로 의심하게 하는 계기가 됩니다.
Q
NMOSD의 시신경염은 MS 관련 시신경염과 어떻게 다른가요?
3. 원인 및 위험 요인
섹션 제목: “3. 원인 및 위험 요인”NMOSD의 정확한 병인은 완전히 밝혀지지 않았습니다. 자가면역 관용의 상실이 근본적인 원인으로 생각됩니다.
주요 위험 요인은 다음과 같습니다.
- 여성: 남녀 비율 약 1:9로 압도적으로 여성에 많습니다.
- 인종: 아시아계 및 아프리카계에서 발병 위험이 높습니다.
- 자가면역 질환 동반: 전신성 홍반성 루푸스(SLE), 쇼그렌 증후군, 중증 근무력증 등
- NMOSD의 10~30%에서 쇼그렌 증후군이 동반됩니다. 소아 사례에서도 보고되었습니다5)
- 중증 근무력증과의 동반은 2~3%에서 관찰됩니다1)
- 악성 종양(부종양성 NMOSD): NMOSD의 3~5%가 부종양성으로 추정됩니다2)3)
- 유방암, 폐암, 난소 기형종 등이 보고되었습니다3)
- 종양 내 AQP4 발현이 자가면역 반응을 유발한다는 기전이 제안되었습니다2)
- 기형종 관련 NMOSD는 젊은 여성(평균 32.7세)에서 많습니다2)
4. 진단 및 검사 방법
섹션 제목: “4. 진단 및 검사 방법”진단 기준 (2015년 국제 컨센서스)
섹션 제목: “진단 기준 (2015년 국제 컨센서스)”AQP4-IgG 양성 NMOSD의 진단 기준은 다음 세 항목을 충족해야 합니다.
- 적어도 하나의 주요 임상 특징
- AQP4-IgG 양성 (최상의 검출 방법 사용)
- 다른 진단의 배제
AQP4-IgG 음성 또는 미검사 NMOSD의 진단 기준은 다음 네 항목을 충족해야 합니다.
- 적어도 두 개의 주요 임상 특징 (그중 하나는 시신경염, LETM 또는 마지막구역 증후군이어야 함)
- 공간적 다발성
- 추가 MRI 요건 충족
- 다른 진단 배제
**주요 임상 특징(6항목)**은 다음과 같습니다:
- 시신경염
- 급성 척수염
- 마지막구역 증후군(난치성 딸꾹질, 메스꺼움 및 구토)
- 급성 뇌간 증후군
- 증상성 기면증/급성 간뇌 증후군
- 증상성 대뇌 증후군
혈청학적 검사
섹션 제목: “혈청학적 검사”아래 표는 주요 항체 검사 방법의 비교를 보여줍니다.
| 검사 방법 | 민감도 | 특이도 | 비고 |
|---|---|---|---|
| CBA (세포 기반 검사) | 69.7~100% | 85.8~100% | 권장 방법 |
| ELISA법 | CBA보다 약간 낮음 | CBA보다 약간 낮음 | 일본에서 보험 적용 |
- AQP4-IgG: NMOSD에 질환 특이적입니다. 급성 발작 시 및 면역억제 치료 시작 전 측정이 권장됩니다1)
- CBA (세포 기반 검사): 현재 권장되는 검출 방법입니다. ELISA의 위양성률은 CBA의 5배로 보고됩니다1)
- MOG-IgG: AQP4-IgG 음성 NMOSD의 약 30%에서 양성입니다1)
- 뇌척수액 올리고클론 밴드 (OCB): NMOSD에서는 10~20%로 낮은 비율(MS에서는 88%). 음성은 NMOSD를 시사합니다1)
- 뇌척수액 백혈구 수: 50/μL 초과, 호중구 또는 호산구의 존재는 NMOSD를 MS와 구별하는 단서가 됩니다
- 임계 깜빡임 빈도 (CFF): 시신경염의 활동성 평가에 유용하며, NMOSD에서 감소합니다
영상 진단 (MRI)
섹션 제목: “영상 진단 (MRI)”- 척수 MRI: LETM이 가장 특징적입니다. 중심 회백질 우세. 척수 부종, T1 저신호, Gd 조영증강을 동반합니다. AQP4+ NMOSD의 약 85%가 급성 척수염 시 LETM을 보입니다 1)
- 시신경 MRI: 지방 억제 영상이 필수입니다. 양측성, 긴 구간 염증(50% 이상)이 특징입니다. 후방 부분 및 시교차 침범이 AQP4+ NMOSD에 특징적입니다 1)
- 뇌 MRI: 마지막구역 병변, 제4뇌실 주위 뇌간 병변, 시상하부/제3뇌실 주위 병변, 광범위 백질 병변 등이 관찰될 수 있습니다
- NMOSD의 특징: MS와 달리 무증상의 새로운 T2 병변은 드뭅니다(3~13%). 감시 MRI는 일반적으로 필요하지 않습니다 1)
감별 진단
섹션 제목: “감별 진단”- 다발성 경화증(MS)
- MOG 항체 관련 질환(MOGAD)
- 급성 파종성 뇌척수염(ADEM)
- 전신성 홍반성 루푸스
- 신경 베체트병
- 고령 환자에서는 허혈성 시신경병증, 경추증성 척수병증, 척수 경색, 원발성 중추신경계 림프종과의 감별이 중요합니다
Q
AQP4 항체가 음성이어도 NMOSD로 진단될 수 있나요?
A
네. AQP4-IgG 음성이거나 검사하지 않은 경우에도, 두 가지 이상의 주요 임상 특징을 충족하고 추가 MRI 요건을 만족하며 다른 질환을 배제하면 NMOSD로 진단됩니다. 또한 AQP4-IgG 음성 사례의 약 30%에서 MOG-IgG가 양성이므로 두 항체 검사가 권장됩니다. AQP4-IgG 음성 사례 중 이후 혈청전환되는 경우는 1% 미만으로 알려져 있습니다.
5. 표준 치료법
섹션 제목: “5. 표준 치료법”급성기 치료
섹션 제목: “급성기 치료”1차 선택: 스테로이드 펄스 요법
- 메틸프레드니솔론 1,000mg/일 정맥 점적 주입을 3일간 시행
- 시력 호전이 없으면 3~4일 간격을 두고 다시 1코스 시행을 고려
- NMOSD의 시신경염은 스테로이드에 대한 내성이 높으므로, 반응이 불충분하면 조기에 다음 치료를 고려
2차 선택: 혈장 교환 요법
스테로이드 펄스에 반응하지 않는 경우 시행한다. 다음 방법이 선택지가 된다.
- 단순 혈장 교환(PE): 효과는 가장 높지만 신체 손상도 최대
- 이중막 여과 혈장 교환(DFPP)
- 면역 흡착 요법(IA): 항체 선택적 제거 가능
효과 순서는 단순 혈장 교환 > 이중막 여과 > 면역 흡착으로 알려져 있다. 1코스 5~6회 시행하며, 치료 후 체내 IgG 양이 회복될 때까지 입원 관리가 필요하다. 참고로 ‘시신경염’에 대해서는 보험 적용이 되지 않는 경우가 있어 환자에게 설명이 필요하다.
재발 예방(유지 요법)
섹션 제목: “재발 예방(유지 요법)”AQP4+ NMOSD에서는 첫 발작 후 유지 치료를 조기에 시작해야 한다고 알려져 있다1). 혈장 교환 후 프레드니솔론 510mg/일 + 아자티오프린 50100mg/일로 전환하는 것이 일반적이다.
근거 수준이 높은 생물학적 제제는 다음과 같다.
보체 억제제
- 에쿨리주맙: 900mg IV 매주 × 4회 → 1,200mg 2주마다 유지 투여1)
- 라불리주맙: 체중 기반 부하 용량(2,400–3,000 mg) → 15일째부터 8주마다 3,000–3,600 mg1)
B세포 제거 요법
- 리툭시맙: 375 mg/m² 정맥주사, 매주 ×4회, 또는 1,000 mg ×2회(2주 간격) → 6개월마다 1,000 mg ×2회1)
- 이네빌리주맙: 300 mg 정맥주사, 15일 간격으로 2회 → 6개월마다1)
IL-6 수용체 억제제
- 사트랄리주맙: 120 mg 피하주사, 4주마다1)
Q
스테로이드 펄스 요법이 효과가 없으면 어떻게 합니까?
A
혈장교환술이 다음 선택지가 됩니다. 단순 혈장교환, 이중막 여과 혈장교환, 면역흡착의 세 가지 중에서 선택합니다. 단순 혈장교환이 가장 효과가 높다고 알려져 있지만 신체에 대한 부담도 큽니다. 1쿠르(5~6회) 시행하며, 치료 후에는 입원 관리가 필요합니다.
6. 병태생리학·상세한 발병 기전
섹션 제목: “6. 병태생리학·상세한 발병 기전”NMOSD는 본질적으로 별아교세포병(astrocytopathy)입니다. 발병 기전은 다음과 같습니다.
항체 생성과 혈액뇌장벽(BBB) 통과
말초에서 B 세포는 AQP4-IgG를 분비하는 형질모세포로 분화합니다. IL-6는 이 분화를 촉진하고 혈액뇌장벽 투과성을 증가시킵니다. 마지막구역(area postrema)은 혈액뇌장벽이 없는 영역으로, AQP4-IgG가 중추신경계로 침투하는 경로가 될 수 있습니다.
별아교세포 손상의 연쇄반응
AQP4-IgG는 별아교세포 족돌기에 고밀도로 발현된 AQP4 수분 채널에 결합하여 다음 경로를 통해 별아교세포 손상을 유발합니다.
- 보체 고전 경로 활성화: AQP4-IgG의 Fc 부분이 보체를 활성화하여 막공격복합체(MAC)를 형성하고 별아교세포를 직접 손상시킵니다.
- 항체의존성 세포매개 세포독성(ADCC): NK 세포와 호중구가 Fcγ 수용체를 통해 별아교세포를 손상시킵니다.
- C5a 아니필라톡신 방출: 과립구(호중구, 호산구)를 동원하여 이차적인 축삭 손상과 탈수초를 유발합니다1).
다발성경화증과의 병리학적 차이
다발성경화증에서는 CD8+ T 세포가 중심 역할을 하며 백질 중심의 탈수초가 주를 이루지만, NMOSD에서는 CD4+ T 세포의 관여가 크고 회백질과 백질 모두에 걸친 괴사성 병변을 형성합니다.
분포 이유
AQP4 채널은 시신경, 마지막구역, 척수에 많이 분포되어 있어 이 영역들이 선택적으로 표적이 됩니다.
바이오마커
- 혈청 GFAP: 별아교세포 손상을 반영하며 발작 시 높은 값을 보입니다.
- 혈청 신경섬유경쇄(NfL): 축삭 손상을 반영하며 발작 중증도와 상관관계가 있습니다1).
관여하는 사이토카인으로 IL-6, IL-10, IL-17a, G-CSF, TNF-α, BAFF/APRIL이 보고되었습니다.
7. 최신 연구와 향후 전망 (연구 단계 보고)
섹션 제목: “7. 최신 연구와 향후 전망 (연구 단계 보고)”부종양성 NMOSD의 기전 규명과 종양 선별검사
섹션 제목: “부종양성 NMOSD의 기전 규명과 종양 선별검사”NMOSD의 3~5%가 부종양성으로 추정됩니다. 난소 기형종과 관련된 사례가 특히 자세히 연구되었습니다.
Ikeguchi 등(2021)은 난소 기형종 관련 AQP4+ NMOSD 6예에 대한 리뷰를 수행했습니다2). 모든 환자는 여성이었고, 평균 발병 연령은 32.7세(15~50세)였습니다. 6예 중 83%(5/6)가 오심과 구토를 보였고, 83%에서 CSF 올리고클론 밴드 양성, 83%에서 등쪽 뇌간 병변이 확인되었습니다. 병리 분석에서는 종양 내 GFAP 양성 신경 조직에 AQP4 면역반응성과 림프구 침윤이 확인되어, 종양 내 AQP4 항원 제시가 자가면역 반응을 유발하는 기전이 시사되었습니다. 종양 절제 후 60%(3/5)에서 AQP4-IgG가 음전되었습니다.
Ding 등(2021)은 43예의 부종양성 NMOSD에 대한 리뷰를 수행했습니다3). 88.4%가 여성이었고, 유방암과 폐암이 가장 흔한 종양 유형이었습니다. 특히 50세 이상의 NMOSD 환자에서 종양 선별검사의 중요성이 강조됩니다.
젊은 환자에서도 기형종을 포함한 종양 선별검사가 권장됩니다.
난치성 사례에 대한 면역흡착 요법
섹션 제목: “난치성 사례에 대한 면역흡착 요법”스테로이드, 혈장교환, 리툭시맙 등에 불응성인 난치성 NMOSD에 대해 Protein-A 면역흡착 요법(IA)의 유효성이 보고되었습니다.
Fan 등(2024)은 스테로이드 펄스 및 IVIG에 반응하지 않는 난치성 쇼그렌 증후군 동반 NMOSD를 앓는 35세 여성에게 Protein-A 면역흡착 3회 세션을 시행했습니다4). 1주일 이내에 시력 장애, 하반신 마비, 고유감각 장애가 현저히 개선되었고, AQP4-IgG, IgA, IgG, IgM의 급속한 감소가 확인되었습니다. 4년간의 추적 관찰에서 재발이나 진행은 관찰되지 않았습니다.
자가면역 질환 동반 사례의 특징
섹션 제목: “자가면역 질환 동반 사례의 특징”NMOSD에 다양한 자가면역 질환이 동반되는 실제 양상이 밝혀지고 있습니다.
Zhu 등(2025)은 11세에 NMOSD가 발병한 14세 여아의 증례를 보고했습니다5). AQP4-IgG 양성이었고, 경과 중 원발성 쇼그렌 증후군의 동반이 확인되었습니다. 메틸프레드니솔론, IVIG, 미코페놀레이트 모페틸(MMF)에서 타크로리무스로의 전환을 통해 관해를 유지하고 있습니다. 성인 NMOSD 증례의 20~30%에서 자가면역 질환 동반이 보고되었지만, 소아 증례에서도 동반이 존재할 수 있음이 제시되었습니다5).
8. 참고문헌
섹션 제목: “8. 참고문헌”- Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
- Ikeguchi R, Shimizu Y, Shimomura A, Suzuki M, Shimoji K, Motohashi T, Yamamoto T, Shibata N, Kitagawa K. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045. doi:10.1212/NXI.0000000000001045. PMID: 34285095. PMCID: PMC8293286.
- Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain and behavior. 2021;11(10):e2282. doi:10.1002/brb3.2282. PMID:34520629; PMCID:PMC8553315.
- Fan W, Chen X, Xiao P, Wei B, Zhang Y, Huang J, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Frontiers in immunology. 2024;15:1429405. doi:10.3389/fimmu.2024.1429405. PMID:39055718; PMCID:PMC11269126.
- Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.