視神経炎

視神経脊髄炎(NMOSD)
1. 視神経脊髄炎(NMOSD)とは
Section titled “1. 視神経脊髄炎(NMOSD)とは”視神経脊髄炎スペクトラム障害(NMOSD)は、中枢神経系を侵す炎症性・抗体介在性・自己免疫疾患である。以前は「デビック病」とも呼ばれ、長年にわたり多発性硬化症(MS)の亜型と見なされてきた。しかし2004年にアクアポリン4(AQP4)に対する自己抗体(AQP4-IgG)が発見されたことで、独立した疾患単位として確立した。
歴史的背景として、1870年にSir Thomas Clifford Allbuttが脊髄炎と視神経障害の関連性を初めて記述した。その後の研究でMSとは異なる疾患と認識され、現在では「NMOSD」という包括的概念で捉えられている。
疫学は以下の通りである。
- 発生率:AQP4+NMOSDの推定年間発生率は0.4〜7.3/百万人1)
- 性差:男女比は約1:9と女性に著しく偏る
- 発症年齢:主に中年(40〜60歳)に好発。本邦では30歳代後半〜40歳代前半にピーク
- 人種:アフリカ系・アジア系に多い傾向1)
- 妊娠との関連:女性の約20〜47%が妊娠中または出産・流産から1年以内に初発する
Q
NMOSDと多発性硬化症(MS)はどう違いますか?
2. 主な症状と臨床所見
Section titled “2. 主な症状と臨床所見”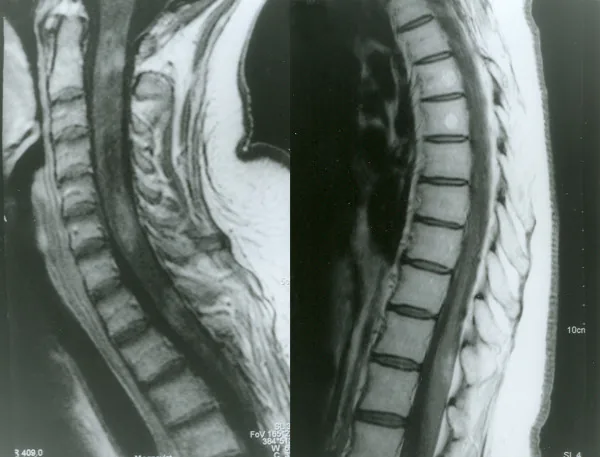
Petelin Gadze Z, et al. Patient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case report. BMC Neurol. 2009. Figure 2. PMCID: PMC2773232. License: CC BY.
2007年4月に撮影された脊髄のガドリニウム造影T1強調MRI像であり、C2、C4-C5、およびTh1レベルに不均一な環状の造影増強効果が認められる。本文「2. 主な症状と臨床所見」の項で扱う脊髄炎に対応する。
NMOSDの症状は侵される部位によって多様である。
- 急激な視力低下:主要症状のひとつ。ステロイド治療への抵抗性が特徴的である
- 眼痛:視神経炎に伴い、約半数の症例に認められる
- 色覚異常:赤色飽和度の低下が典型的
- 視野障害:中心暗点にとどまらず、水平半盲・両耳側半盲・同名半盲をきたすこともある。視交叉・視索にまで病変が及ぶためである
- 感覚障害・対麻痺:脊髄炎による運動・感覚障害
- 膀胱直腸障害:脊髄炎に伴う自律神経障害
- 難治性吃逆・悪心嘔吐:最後野(area postrema)の病変による特徴的症状
- 眼球運動障害:脳幹病変によるもの
- 過眠症(ナルコレプシー様):間脳・視床下部病変による
発症初期にインフルエンザ様症状(発熱・筋肉痛・頭痛)を呈することがある。
脊髄炎
最後野症候群
Q
NMOSDの視神経炎はMS関連のものとどう違いますか?
3. 原因とリスク要因
Section titled “3. 原因とリスク要因”NMOSDの正確な病因は完全には解明されていない。自己免疫寛容の喪失が根本にあると考えられている。
主なリスク要因は以下の通りである。
- 女性:男女比約1:9と圧倒的に女性に多い
- 人種:アジア系・アフリカ系で発症リスクが高い
- 自己免疫疾患の合併:全身性エリテマトーデス(SLE)、シェーグレン症候群、重症筋無力症など
- NMOSDの10〜30%にシェーグレン症候群が合併する。小児例でも報告されている5)
- 重症筋無力症との合併は2〜3%に認められる1)
- 悪性腫瘍(傍腫瘍性NMOSD):NMOSDの3〜5%が傍腫瘍性と推定される2)3)
- 乳癌・肺癌・卵巣奇形腫などが報告されている3)
- 腫瘍内のAQP4発現が自己免疫反応を誘発するという機序が提唱されている2)
- 奇形腫関連NMOSDは若年女性(平均32.7歳)に多い2)
4. 診断と検査方法
Section titled “4. 診断と検査方法”診断基準(2015年国際コンセンサス)
Section titled “診断基準(2015年国際コンセンサス)”AQP4-IgG陽性NMOSDの診断基準は以下の3項目を満たすことである。
- 少なくとも1つの主要臨床的特徴
- AQP4-IgG陽性(最善の検出法使用)
- 他の診断の除外
AQP4-IgG陰性または未検査のNMOSDの診断基準は以下の4項目を満たすことである。
- 少なくとも2つの主要臨床的特徴(うち1つは視神経炎・LETM・または最後野症候群)
- 空間的多発性
- 追加MRI要件を満たす
- 他の診断の除外
**主要臨床的特徴(6項目)**は以下の通りである。
- 視神経炎
- 急性脊髄炎
- 最後野症候群(難治性吃逆・悪心嘔吐)
- 急性脳幹症候群
- 症候性ナルコレプシー/急性間脳症候群
- 症候性大脳症候群
血清学的検査
Section titled “血清学的検査”以下の表に主要な抗体検査法の比較を示す。
| 検査法 | 感度 | 特異度 | 備考 |
|---|---|---|---|
| CBA(細胞ベースアッセイ) | 69.7〜100% | 85.8〜100% | 推奨法 |
| ELISA法 | CBAよりやや劣る | CBAよりやや劣る | 日本で保険収載 |
- AQP4-IgG:NMOSDに疾患特異的。急性発作時・免疫抑制療法開始前の測定が推奨される1)
- CBA(細胞ベースアッセイ):現在の推奨検出法。ELISAの偽陽性率はCBAの5倍とされる1)
- MOG-IgG:AQP4-IgG陰性NMOSDの約30%で陽性1)
- CSFオリゴクローナルバンド(OCB):NMOSDでは10〜20%と低率(MSでは88%)。陰性はNMOSDを示唆する1)
- CSF白血球数:50/μL超・好中球・好酸球の存在はNMOSDをMSから区別する手がかりとなる
画像診断(MRI)
Section titled “画像診断(MRI)”- 脊髄MRI:LETMが最も特徴的。中央灰白質優位。脊髄腫脹・T1低信号・Gd造影効果を伴う。AQP4+NMOSDの約85%が急性脊髄炎時にLETMを呈する1)
- 視神経MRI:脂肪抑制画像が必須。両側性・長範囲の炎症(50%以上)が特徴。後方部分・視交叉の関与がAQP4+NMOSDに特徴的1)
- 脳MRI:最後野病変・第4脳室周囲脳幹病変・視床下部/第3脳室周囲病変・広範白質病変などを認める
- NMOSDの特徴:MSとは異なり、無症候性の新規T2病変は稀(3〜13%)。サーベイランスMRIは通常不要1)
- 多発性硬化症(MS)
- MOG抗体関連疾患(MOGAD)
- 急性散在性脳脊髄炎(ADEM)
- 全身性エリテマトーデス
- 神経ベーチェット病
- 高齢者では虚血性視神経症・頸椎症性脊髄症・脊髄梗塞・原発性CNSリンパ腫との鑑別が重要
Q
AQP4抗体が陰性でもNMOSDと診断されることはありますか?
5. 標準的な治療法
Section titled “5. 標準的な治療法”第一選択:ステロイドパルス療法
- メチルプレドニゾロン1,000mg/日の点滴静注を3日間施行する
- 視力改善がなければ中3〜4日あけて再度1クール施行を検討する
- NMOSDの視神経炎はステロイドへの抵抗性が高いため、反応不十分な場合は早期に次の治療を考慮する
第二選択:血漿交換療法
ステロイドパルスに反応しない場合に実施する。以下の方法が選択肢となる。
- 単純血漿交換療法(PE):効果は最も高いが生体へのダメージも最大
- 二重膜濾過血漿交換療法(DFPP)
- 免疫吸着療法(IA):抗体選択的除去が可能
効果の順は単純血漿交換 > 二重膜濾過 > 免疫吸着とされる。1クール5〜6回施行し、治療後は体内IgG量の回復まで入院管理が必要である。なお「視神経炎」に対しては保険適用外となる場合があり、患者への説明が必要である。
再発予防(維持療法)
Section titled “再発予防(維持療法)”AQP4+NMOSDでは初回発作後から維持治療を早期開始すべきとされる1)。日本では血漿交換後にプレドニゾロン5〜10mg/日+アザチオプリン50〜100mg/日への移行が一般的である。
エビデンスレベルの高い生物学的製剤は以下の通りである。
補体阻害薬
- エクリズマブ:900mg IV 毎週×4回→1,200mg 2週間ごとの維持投与1)
- ラブリズマブ:体重ベースの負荷量(2,400〜3,000mg)→15日目以降3,000〜3,600mg を8週ごと1)
B細胞除去療法
- リツキシマブ:375mg/m² IV 毎週×4回、または1,000mg×2回(2週間隔)→6か月ごとに1,000mg×2回1)
- イネビリズマブ:300mg IV 15日ごとに2回→6か月ごと1)
IL-6受容体阻害薬
- サトラリズマブ:120mg 皮下注射、4週間ごと1)
Q
ステロイドパルス療法が効かない場合はどうしますか?
A
血漿交換療法が次の選択肢となる。単純血漿交換・二重膜濾過血漿交換・免疫吸着の3種類から選択する。単純血漿交換が最も効果が高いとされるが生体への負担も大きい。1クール5〜6回施行し、治療後は入院管理が必要となる。
6. 病態生理学・詳細な発症機序
Section titled “6. 病態生理学・詳細な発症機序”NMOSDは本質的にはアストロサイト病(astrocytopathy)である。発症機序は以下の通りである。
抗体産生と血液脳関門(BBB)通過
末梢でB細胞がAQP4-IgG分泌プラスマブラストに分化する。IL-6はこの分化を促進し、BBBの透過性を亢進させる。最後野はBBBを欠く領域であり、AQP4-IgGがCNSへ侵入する経路となりうる。
アストロサイト障害の連鎖
アストロサイト足突起に高密度に発現するAQP4水チャネルにAQP4-IgGが結合し、以下の経路でアストロサイト傷害が生じる。
- 補体古典経路の活性化:AQP4-IgGのFc部分が補体を活性化し、膜傷害複合体(MAC)を形成してアストロサイトを直接傷害する
- ADCC(抗体依存性細胞傷害):NK細胞や好中球がアストロサイトをFcγ受容体を介して傷害する
- C5aアナフィラトキシン放出:顆粒球(好中球・好酸球)を動員し、二次的な軸索損傷と脱髄を引き起こす1)
MSとの病理的差異
MSではCD8+ T細胞が中心的役割を担う白質中心の脱髄が主体であるが、NMOSDではCD4+ T細胞の関与が大きく、灰白質・白質の両方に及ぶ壊死性病変を形成する。
分布の理由
AQP4チャネルは視神経・最後野・脊髄に多く分布しており、これらの領域が選択的に標的となる。
バイオマーカー
- 血清GFAP:アストロサイト障害を反映し、発作時に高値となる
- 血清ニューロフィラメント軽鎖(NfL):軸索損傷を反映し、発作の重症度と相関する1)
関与するサイトカインとして IL-6・IL-10・IL-17a・G-CSF・TNF-α・BAFF/APRIL が報告されている。
7. 最新の研究と今後の展望(研究段階の報告)
Section titled “7. 最新の研究と今後の展望(研究段階の報告)”傍腫瘍性NMOSDの機序解明と腫瘍スクリーニング
Section titled “傍腫瘍性NMOSDの機序解明と腫瘍スクリーニング”NMOSDの3〜5%が傍腫瘍性と推定される。卵巣奇形腫との関連例が特に詳しく研究されている。
Ikeguchiら(2021)は卵巣奇形腫関連AQP4+NMOSDの6例レビューを実施した2)。全例女性、平均発症年齢32.7歳(15〜50歳)。6例のうち83%(5/6例)が悪心・嘔吐を呈し、83%でCSFオリゴクローナルバンド陽性、83%に背側脳幹病変を認めた。病理解析では腫瘍内のGFAP陽性神経組織にAQP4免疫反応性とリンパ球浸潤が確認され、腫瘍内でのAQP4抗原提示が自己免疫反応を惹起する機序が示唆された。腫瘍摘出後には60%(3/5例)でAQP4-IgGが陰転化した。
Dingら(2021)は43例の傍腫瘍性NMOSDのレビューを実施した3)。88.4%が女性で、乳癌と肺癌が最多の腫瘍型であった。特に50歳以上のNMOSD患者では腫瘍スクリーニングの重要性が強調されている。
若年例であっても奇形腫を含む腫瘍のスクリーニングが推奨される。
難治例に対する免疫吸着療法
Section titled “難治例に対する免疫吸着療法”ステロイド・血漿交換・リツキシマブなどに不応性の難治性NMOSDに対して、Protein-A免疫吸着療法(IA)の有効性が報告されている。
Fanら(2024)は、ステロイドパルスおよびIVIGに反応しない難治性シェーグレン症候群合併NMOSDの35歳女性に対し、Protein-A免疫吸着療法3セッションを実施した4)。1週間以内に視力障害・対麻痺・固有感覚障害が著明に改善し、AQP4-IgG・IgA・IgG・IgMの急速な低下が確認された。4年間の経過観察で再発・進行は認めていない。
自己免疫疾患との合併例の特徴
Section titled “自己免疫疾患との合併例の特徴”NMOSDへの各種自己免疫疾患合併の実態が明らかになりつつある。
Zhuら(2025)は、11歳時にNMOSDを発症した14歳女児の症例を報告した5)。AQP4-IgG陽性で、経過中に原発性シェーグレン症候群の合併が確認された。メチルプレドニゾロン・IVIG・ミコフェノール酸モフェチル(MMF)→タクロリムスへの変更により寛解を維持している。成人NMOSD症例の20〜30%に自己免疫疾患合併が報告されているが、小児例での合併も存在することが示された5)。
8. 参考文献
Section titled “8. 参考文献”- Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment: A Tale of Two Central Nervous System Autoimmune Inflammatory Disorders. Neurol Clin. 2024;42(1):77-114. doi:10.1016/j.ncl.2023.06.009. PMID:37980124; PMCID:PMC10658081.
- Ikeguchi R, Shimizu Y, Shimomura A, Suzuki M, Shimoji K, Motohashi T, Yamamoto T, Shibata N, Kitagawa K. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045. doi:10.1212/NXI.0000000000001045. PMID: 34285095. PMCID: PMC8293286.
- Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain and behavior. 2021;11(10):e2282. doi:10.1002/brb3.2282. PMID:34520629; PMCID:PMC8553315.
- Fan W, Chen X, Xiao P, Wei B, Zhang Y, Huang J, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Frontiers in immunology. 2024;15:1429405. doi:10.3389/fimmu.2024.1429405. PMID:39055718; PMCID:PMC11269126.
- Zhu GQ, Hu RX, Peng Y, Yao Y, Li GM. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Frontiers in immunology. 2025;16:1559825. doi:10.3389/fimmu.2025.1559825. PMID:40698091; PMCID:PMC12279711.