โรคกลุ่มอาการอักเสบของเส้นประสาทตา และไขสันหลัง (NMOSD ) เป็นโรคภูมิคุ้มกันทำลายตนเองของระบบประสาทส่วนกลางที่เกิดจากแอนติบอดีต่ออะควาพอริน 4 (AQP4) เป็นหลัก
อาการหลักคือเส้นประสาทตา อักเสบและไขสันหลังอักเสบ พบมากในผู้หญิง (อัตราส่วนหญิง:ชายประมาณ 9:1) วัยกลางคน และเชื้อสายเอเชีย
โรคเส้นประสาทตา อักเสบใน NMOSD รุนแรงกว่า เป็นสองข้าง และกลับเป็นซ้ำบ่อยกว่าในโรคปลอกประสาทเสื่อมแข็ง (MS ) โดยมีโอกาสเกิดความบกพร่องทางการมองเห็น ถาวรที่ระดับ 20/200 หรือแย่กว่าในตาข้างเดียวสูงถึง 60-69%ในโรคไขสันหลังอักเสบ (myelitis) ลักษณะเด่นคือการอักเสบตามยาวของไขสันหลัง (LETM) ที่กินระยะตั้งแต่ 3 กระดูกสันหลังขึ้นไป และทำให้เกิดกลุ่มอาการไขสันหลังเสียการทำงานโดยสมบูรณ์ (ความผิดปกติด้านการเคลื่อนไหว ความรู้สึก และระบบประสาทอัตโนมัติ)
การวินิจฉัยอาศัยการตรวจ AQP4-IgG (แนะนำใช้วิธี cell-based assay) และ MRI เป็นหลัก
ในระยะเฉียบพลันให้การรักษาด้วยสเตียรอยด์ แบบ pulse หากตอบสนองไม่ดี ทางเลือกถัดไปคือการแลกเปลี่ยนพลาสมา (plasmapheresis)
การป้องกันการกลับเป็นซ้ำใช้ยาต้านคอมพลีเมนต์ (eculizumab), การกำจัดเซลล์ B (rituximab), หรือยาต้าน IL-6 receptor (satralizumab) และแนะนำให้เริ่มการรักษาตั้งแต่หลังการเกิดอาการครั้งแรก
โรคปลอกประสาทอักเสบเส้นประสาทตา และไขสันหลัง (NMOSD ) เป็นโรคอักเสบที่เกิดจากแอนติบอดีและภูมิคุ้มกันทำลายตนเอง ซึ่งส่งผลต่อระบบประสาทส่วนกลาง เดิมเรียกว่า “โรคเดวิก” และถูกมองว่าเป็นชนิดย่อยของโรคปลอกประสาทเสื่อมแข็ง (MS ) มานานหลายปี แต่ในปี 2004 การค้นพบแอนติบอดีต่อตนเองต่ออะควาพอริน 4 (AQP4-IgG) ทำให้โรคนี้ถูกจัดเป็นโรคอิสระ
ภูมิหลังทางประวัติศาสตร์ ในปี 1870 เซอร์โทมัส คลิฟฟอร์ด ออลบัตต์ เป็นคนแรกที่อธิบายความสัมพันธ์ระหว่างไขสันหลังอักเสบและความผิดปกติของเส้นประสาทตา การวิจัยในภายหลังทำให้โรคนี้ถูกแยกออกจาก MS และปัจจุบันจัดอยู่ในแนวคิดรวมที่เรียกว่า “NMOSD ”
ระบาดวิทยา มีดังนี้
อุบัติการณ์ : อุบัติการณ์รายปีโดยประมาณของ AQP4+NMOSD อยู่ที่ 0.4 ถึง 7.3 ต่อล้านคน1) ความแตกต่างทางเพศ : อัตราส่วนชายต่อหญิงประมาณ 1:9 โดยพบในผู้หญิงมากกว่าอย่างชัดเจนอายุที่เริ่มมีอาการ : มักพบในวัยกลางคน (40-60 ปี) ในญี่ปุ่นพบสูงสุดในช่วงอายุ 30 ปลายถึง 40 ต้นเชื้อชาติ : มักพบในคนเชื้อสายแอฟริกันและเอเชีย1) ความสัมพันธ์กับการตั้งครรภ์ : ผู้หญิงประมาณ 20–47% มีอาการครั้งแรกในระหว่างตั้งครรภ์หรือภายใน 1 ปีหลังคลอดหรือแท้งบุตร
Q
NMOSD กับโรคปลอกประสาทเสื่อมแข็ง (MS) แตกต่างกันอย่างไร?
A
NMOSD เป็นโรคที่เกิดจากแอนติบอดีต่อช่องน้ำ AQP4 บนแอสโทรไซต์ ซึ่งมีพยาธิสภาพ การรักษา และพยากรณ์โรคที่แตกต่างโดยพื้นฐานจาก MS ใน NMOSD มักพบ LETM หรือโรคประสาทอักเสบรุนแรง และยาปรับเปลี่ยนโรคที่ได้ผลใน MS เช่น อินเตอร์เฟอรอนเบตา อาจกระตุ้นให้ NMOSD กำเริบได้ ซึ่งเป็นความแตกต่างที่สำคัญ
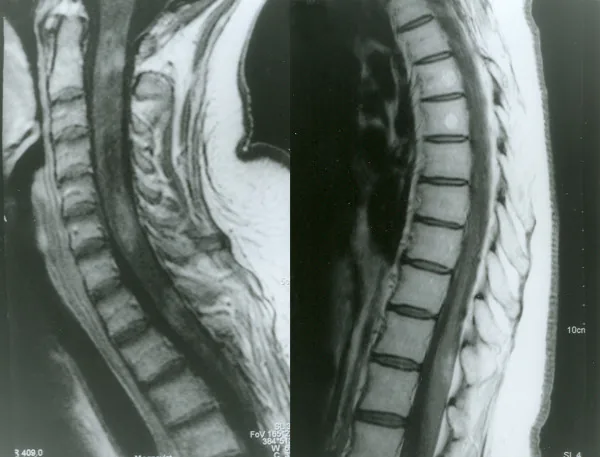
MRI ที่มีการเพิ่มความเข้มของไขสันหลังในโรคประสาทตาอักเสบร่วมกับไขสันหลังอักเสบ Petelin Gadze Z, et al. Patient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case report. BMC Neurol. 2009. Figure 2. PM
CI D: PMC2773232. License: CC BY.
ภาพ MRI ถ่วงน้ำหนัก T1 ที่มีการเพิ่มความเข้มของแกโดลิเนียมของไขสันหลัง ถ่ายเมื่อเดือนเมษายน พ.ศ. 2550 แสดงให้เห็นการเพิ่มความเข้มแบบวงแหวนที่ไม่สม่ำเสมอที่ระดับ C2, C4-C5 และ Th1 ซึ่งสอดคล้องกับไขสันหลังอักเสบที่กล่าวถึงในหัวข้อ “2. อาการหลักและอาการแสดงทางคลินิก”
อาการของ NMOSD มีความหลากหลายขึ้นอยู่กับตำแหน่งที่ถูกกระทบ
การมองเห็น ลดลงอย่างรวดเร็วสเตียรอยด์ ปวดตา ความผิดปกติของการมองเห็นสี ความบกพร่องของลานสายตา จุดบอดกลาง แต่ยังอาจเกิดภาวะ hemianopia แนวราบ, hemianopia ข้างขมับทั้งสองข้าง, หรือ homonymous hemianopia เนื่องจากรอยโรคขยายไปถึง optic chiasm และ optic tractความผิดปกติทางความรู้สึกและอัมพาตครึ่งล่าง : ความผิดปกติทางการเคลื่อนไหวและความรู้สึกจากไขสันหลังอักเสบความผิดปกติของกระเพาะปัสสาวะและลำไส้ : ความผิดปกติของระบบประสาทอัตโนมัติที่เกิดจากไขสันหลังอักเสบอาการสะอึกที่รักษายากและคลื่นไส้อาเจียน : อาการเฉพาะจากรอยโรคที่ area postremaความผิดปกติของการเคลื่อนไหวลูกตา อาการง่วงซึมมากเกินไป (คล้ายเฉียบหลับ) : เกิดจากรอยโรคที่ไดเอนเซฟาลอน/ไฮโปทาลามัส
ในระยะเริ่มแรกของโรค อาจมีอาการคล้ายไข้หวัดใหญ่ (ไข้ ปวดกล้ามเนื้อ ปวดศีรษะ)
โรคประสาทอักเสบตา
ภาวะบวมของจานประสาทตา (Optic disc edema) : พบในระยะเฉียบพลัน ต่อมาจะเปลี่ยนเป็นฝ่อของจานประสาทตา
RAPD (Relative Afferent Pupillary Defect)
เกิดพร้อมกันทั้งสองข้าง : โรคปลอกประสาทอักเสบ NMOSD พบการอักเสบของเส้นประสาทตา พร้อมกันทั้งสองข้างร้อยละ 17-82 ซึ่งเป็นข้อแตกต่างสำคัญจาก MS
การมองเห็น ลดลงอย่างรุนแรงNMOSD ค่ามัธยฐานของระดับการมองเห็น ต่ำสุดอยู่ที่ระดับนับนิ้ว (HM) หลังฟื้นตัว ค่ามัธยฐานยังคงอยู่ที่ระดับนับนิ้ว และร้อยละ 60-69 มีความบกพร่องทางการมองเห็น ถาวรที่ 20/200 หรือแย่กว่าในตาอย่างน้อยหนึ่งข้าง
ไขสันหลังอักเสบ
LETM (ไขสันหลังอักเสบตามยาวกว้าง) : รอยโรคต่อเนื่องยาวตั้งแต่ 3 กระดูกสันหลังขึ้นไป พบในผู้ป่วย AQP4+NMOSD ประมาณ 85% ในช่วงที่มีไขสันหลังอักเสบเฉียบพลัน
กลุ่มอาการไขสันหลังสมบูรณ์ : เกี่ยวข้องกับทั้ง 3 วิถีทาง ได้แก่ การเคลื่อนไหว ความรู้สึก และระบบประสาทอัตโนมัติ
ความบกพร่องทางการทำงานอย่างรุนแรง : ผู้ป่วยมากกว่า 30% ต้องพึ่งพารถเข็นในช่วงที่อาการแย่ที่สุด ผู้ป่วย AQP4+NMOSD ร้อยละ 37–44 ต้องใช้อุปกรณ์ช่วยเดินในที่สุด
กลุ่มอาการอาเรียนหลังสุด
อาการสะอึกที่รักษายาก : เกิดขึ้นต่อเนื่องเป็นเวลาหลายวันถึงหลายสัปดาห์ และไม่ตอบสนองต่อยาแก้อาเจียนทั่วไป
อาการคลื่นไส้ อาเจียน : เนื่องจากบริเวณอาเรียนหลังสุดไม่มีด่านกั้นเลือด-สมอง ทำให้ AQP4-IgG เข้าถึงได้โดยตรง
ลักษณะสำคัญในการวินิจฉัย NMOSD : อาการสะอึกที่รักษายากโดยไม่มีสาเหตุชัดเจนเป็นสัญญาณที่ควรสงสัย NMOSD อย่างยิ่ง
โรคประสาทตาอักเสบ ที่สัมพันธ์กับ MS
โรคประสาทตาอักเสบ ใน NMOSD มักรุนแรง กว้างขวาง เป็นสองข้าง กลับเป็นซ้ำ และฟื้นตัวได้ยากกว่า MS การมีส่วนร่วมของออปติกไคแอสมาคือลักษณะเด่นของ NMOSD ใน MS มักพบจุดบอดกลาง แบบเดี่ยวและการพยากรณ์โรคทางสายตาค่อนข้างดี
Q
โรคประสาทตาอักเสบใน NMOSD แตกต่างจากที่สัมพันธ์กับ MS อย่างไร?
A
โรคประสาทตาอักเสบ ใน NMOSD รุนแรงกว่า เป็นสองข้าง กลับเป็นซ้ำ และมีการพยากรณ์โรคทางสายตาไม่ดี ใน AQP4+ NMOSD พบว่าผู้ป่วย 60-69% มีความบกพร่องทางการมองเห็น ถาวรที่ 20/200 หรือแย่กว่าในตาอย่างน้อยหนึ่งข้าง นอกจากนี้ยังมีแนวโน้มที่จะมีส่วนร่วมของออปติกไคแอสมาทำให้เกิดความบกพร่องของลานสายตา ที่หลากหลาย เช่น hemianopsia แบบสองข้าง ซึ่งแตกต่างจาก MS
ยังไม่ทราบสาเหตุที่แน่ชัดของ NMOSD เชื่อว่ามีสาเหตุจากการสูญเสียความทนทานต่อตนเองของระบบภูมิคุ้มกัน
ปัจจัยเสี่ยงหลัก ได้แก่:
เพศหญิง : พบในผู้หญิงมากกว่าผู้ชายในอัตราส่วนประมาณ 1:9เชื้อชาติ : ความเสี่ยงในการเกิดโรคสูงในชาวเอเชียและแอฟริกาโรคร่วม autoimmune : โรคลูปัส erythematosus ทั่วร่างกาย (SLE ), กลุ่มอาการ Sjögren, โรคกล้ามเนื้ออ่อนแรงชนิดร้ายแรง (myasthenia gravis) เป็นต้น
NMOSD ร่วมกับกลุ่มอาการ Sjögren พบได้ 10–30% มีรายงานในเด็กด้วย5) การร่วมกับโรคกล้ามเนื้ออ่อนแรงชนิดร้ายแรงพบได้ 2–3%1)
เนื้องอกมะเร็ง (NMOSD แบบพารานีโอพลาสติก) : ประมาณ 3-5% ของ NMOSD คาดว่าเป็นแบบพารานีโอพลาสติก2) 3)
มีรายงานมะเร็งเต้านม มะเร็งปอด และเทราโทมาของรังไข่3)
มีการเสนอว่าการแสดงออกของ AQP4 ในเนื้องอกกระตุ้นการตอบสนองทางภูมิคุ้มกันตนเอง2)
NMOSD ที่เกี่ยวข้องกับเทราโทมาพบมากในหญิงสาว (อายุเฉลี่ย 32.7 ปี)2)
หากคุณมีโรคภูมิต้านตนเอง เช่น กลุ่มอาการโจเกรนหรือโรคลูปัส (SLE ) และมีอาการตามัวลงอย่างรวดเร็ว หรือมีอาการสะอึกและอาเจียนที่รักษายาก ควรไปพบแพทย์เฉพาะทางด้านประสาทวิทยาหรือจักษุประสาทวิทยาโดยเร็ว โดยคำนึงถึงโรค NMOSD
เกณฑ์การวินิจฉัย NMOSD ที่มี AQP4-IgG บวก คือต้องมีคุณสมบัติครบ 3 ข้อดังต่อไปนี้
มีลักษณะทางคลินิกหลักอย่างน้อย 1 ข้อ
ผล AQP4-IgG เป็นบวก (โดยใช้วิธีตรวจที่ดีที่สุด)
ไม่พบโรคอื่นที่สามารถอธิบายอาการได้
เกณฑ์การวินิจฉัย NMOSD ที่มีผล AQP4-IgG เป็นลบหรือไม่ได้ตรวจ คือต้องเป็นไปตามข้อ 4 ข้อต่อไปนี้
มีลักษณะทางคลินิกหลักอย่างน้อย 2 ข้อ (หนึ่งในนั้นต้องเป็นโรคประสาทอักเสบตา LETM หรือกลุ่มอาการบริเวณ postrema)
มีการกระจายของรอยโรคในหลายตำแหน่ง (spatial dissemination)
เป็นไปตามข้อกำหนดเพิ่มเติมของ MRI
การแยกโรคอื่นออก
ลักษณะทางคลินิกหลัก (6 ข้อ) มีดังนี้
โรคประสาทอักเสบตา
ไขสันหลังอักเสบเฉียบพลัน
กลุ่มอาการบริเวณสุดท้าย (อาการสะอึก คลื่นไส้ อาเจียนที่รักษายาก)
กลุ่มอาการก้านสมองเฉียบพลัน
โรคลมหลับที่มีสาเหตุ/กลุ่มอาการไฮโปทาลามัสเฉียบพลัน
กลุ่มอาการสมองใหญ่ที่มีสาเหตุ
ตารางด้านล่างแสดงการเปรียบเทียบวิธีการตรวจหาแอนติบอดีหลัก
วิธีการตรวจ ความไว ความจำเพาะ หมายเหตุ CBA (การทดสอบโดยใช้เซลล์เป็นฐาน) 69.7–100% 85.8–100% วิธีที่แนะนำ วิธี ELISA ด้อยกว่า CBA เล็กน้อย ด้อยกว่า CBA เล็กน้อย ครอบคลุมโดยประกันในญี่ปุ่น
AQP4-IgG : เฉพาะโรคสำหรับ NMOSD แนะนำให้ตรวจวัดในช่วงที่มีอาการกำเริบเฉียบพลันหรือก่อนเริ่มการรักษาด้วยยากดภูมิคุ้มกัน1) CBA (การทดสอบโดยใช้เซลล์เป็นฐาน) : วิธีการตรวจที่แนะนำในปัจจุบัน อัตราผลบวกลวงของ ELISA สูงกว่า CBA ถึง 5 เท่า1) MOG-IgG NMOSD ที่มี AQP4-IgG เป็นลบ1) แถบโอลิโกโคลนอลในน้ำไขสันหลัง (OCB) : ใน NMOSD พบได้น้อย 10-20% (ใน MS พบ 88%) ผลลบชี้แนะถึง NMOSD 1) จำนวนเม็ดเลือดขาวในน้ำไขสันหลัง (CSF) : มากกว่า 50/μL การมีนิวโทรฟิลและอีโอซิโนฟิลเป็นข้อบ่งชี้ที่ช่วยแยก NMOSD ออกจาก MS
MRI ไขสันหลัง : LETM เป็นลักษณะที่พบได้บ่อยที่สุด โดยมีรอยโรคที่เนื้อเทาส่วนกลางเป็นหลัก ร่วมกับไขสันหลังบวม สัญญาณ T1 ต่ำ และการเพิ่มสัญญาณหลังฉีดแกโดลิเนียม ผู้ป่วย AQP4+NMOSD ประมาณ 85% มี LETM ในช่วงไขสันหลังอักเสบเฉียบพลัน1) MRI เส้นประสาทตา : จำเป็นต้องใช้ภาพแบบระงับสัญญาณไขมัน ลักษณะเด่นคือการอักเสบแบบสองข้างและยาว (>50%) การมีส่วนร่วมของส่วนหลังและออปติกไคแอสมาพบได้บ่อยใน AQP4+NMOSD 1) MRI สมอง : พบรอยโรคที่ area postrema, รอยโรคที่ก้านสมองรอบช่องที่สี่, รอยโรคที่ไฮโปทาลามัส/รอบช่องที่สาม, และรอยโรคเนื้อขาวแบบกว้างลักษณะของ NMOSD : ต่างจาก MS ตรงที่รอยโรค T2 ใหม่ที่ไม่มีอาการพบได้น้อย (3–13%) โดยทั่วไปไม่จำเป็นต้องติดตาม MRI เป็นประจำ1)
โรคปลอกประสาทเสื่อมแข็ง (MS )
โรคที่เกี่ยวข้องกับแอนติบอดี MOG (MOGAD )โรคสมองและไขสันหลังอักเสบเฉียบพลันแพร่กระจาย (ADEM )
โรคลูปัส erythematosus ทั่วร่างกาย
โรคเบห์เซ็ตทางระบบประสาท
ในผู้สูงอายุ การแยกโรคจาก ischemic optic neuropathy, cervical spondylotic myelopathy, spinal cord infarction และ primary CNS lymphoma มีความสำคัญ
Q
แม้ว่า AQP4 antibody จะเป็นลบ ก็สามารถวินิจฉัย NMOSD ได้หรือไม่?
A
ได้ แม้ AQP4-IgG จะเป็นลบหรือไม่ได้ตรวจ หากมีลักษณะทางคลินิกหลักอย่างน้อย 2 ข้อ ตรงตามข้อกำหนด MRI เพิ่มเติม และสามารถแยกโรคอื่นออกได้ ก็สามารถวินิจฉัย NMOSD ได้ นอกจากนี้ ประมาณ 30% ของผู้ป่วยที่ AQP4-IgG เป็นลบจะมี MOG-IgG เป็นบวก จึงแนะนำให้ตรวจวัดแอนติบอดีทั้งสองชนิด ในผู้ป่วยที่ AQP4-IgG เป็นลบ มีน้อยกว่า 1% ที่จะเกิด seroconversion ในภายหลัง
การรักษาทางเลือกแรก: การให้สเตียรอยด์ แบบพัลส์
ให้ methylprednisolone 1,000 มก./วัน ทางหลอดเลือดดำเป็นเวลา 3 วัน
หากการมองเห็น ไม่ดีขึ้น ให้พิจารณาให้การรักษาซ้ำอีก 1 คอร์ส โดยเว้นระยะ 3-4 วัน
โรคประสาทตาอักเสบ ใน NMOSD มักดื้อต่อสเตียรอยด์ ดังนั้นหากตอบสนองไม่เพียงพอ ควรพิจารณาการรักษาถัดไปโดยเร็ว
ทางเลือกที่สอง: การแลกเปลี่ยนพลาสมา
ดำเนินการเมื่อไม่ตอบสนองต่อการให้สเตียรอยด์ แบบพัลส์ วิธีการต่อไปนี้เป็นทางเลือก:
การแลกเปลี่ยนพลาสมา อย่างง่าย (PE)การบำบัดด้วยการแลกเปลี่ยนพลาสมา ผ่านเยื่อกรองสองชั้น (DFPP) การบำบัดด้วยการดูดซับภูมิคุ้มกัน (IA) : สามารถกำจัดแอนติบอดีแบบเลือกสรรได้
ลำดับประสิทธิผลคือ การแลกเปลี่ยนพลาสมา อย่างง่าย > การกรองสองชั้น > การดูดซับภูมิคุ้มกัน โดยทั่วไปจะทำ 5-6 ครั้งต่อคอร์ส หลังการรักษาต้องอยู่โรงพยาบาลเพื่อดูแลจนกว่าระดับ IgG ในร่างกายจะฟื้นตัว สำหรับ “โรคประสาทอักเสบตา” อาจไม่ครอบคลุมในสิทธิประโยชน์ และจำเป็นต้องอธิบายให้ผู้ป่วยทราบ
ใน AQP4+NMOSD ควรเริ่มการรักษาแบบประคับประคองตั้งแต่เนิ่นๆ หลังจากการโจมตีครั้งแรก 1) ในญี่ปุ่น หลังจากการแลกเปลี่ยนพลาสมา มักจะเปลี่ยนไปใช้เพรดนิโซโลน 5-10 มก./วัน ร่วมกับอะซาไธโอพรีน 50-100 มก./วัน
ยาชีวภาพที่มีระดับหลักฐานสูง มีดังนี้
ยาที่ยับยั้งคอมพลีเมนต์
อีคูลิซูแมบ : 900 มก. IV ทุกสัปดาห์ × 4 ครั้ง → การให้ยาบำรุงรักษา 1,200 มก. ทุก 2 สัปดาห์ 1) ลาบลิซูแมบ : ขนาดยาเริ่มต้นตามน้ำหนักตัว (2,400–3,000 มก.) → หลังจากวันที่ 15 ให้ 3,000–3,600 มก. ทุก 8 สัปดาห์1)
การบำบัดด้วยการกำจัดเซลล์บี
ริตูซิแมบ : 375 มก./ตร.ม. ฉีดเข้าเส้นเลือดดำทุกสัปดาห์ ×4 ครั้ง หรือ 1,000 มก. ×2 ครั้ง (ห่างกัน 2 สัปดาห์) → จากนั้น 1,000 มก. ×2 ครั้ง ทุก 6 เดือน1) อินิบิลิซูแมบ : 300 มก. ฉีดเข้าเส้นเลือดดำ 2 ครั้ง ห่างกัน 15 วัน → จากนั้นทุก 6 เดือน1)
ยับยั้งตัวรับ IL-6
ซาทราลิซูแมบ : 120 มก. ฉีดใต้ผิวหนัง ทุก 4 สัปดาห์1)
อินเตอร์เฟียรอนเบตา, นาทาลิซูแมบ และฟิงโกลิโมดที่ใช้ใน MS อาจกระตุ้นหรือทำให้ NMOSD กำเริบขึ้นได้ จึงควรถือเป็นข้อห้ามใช้
ยาป้องกันการกลับเป็นซ้ำจำเป็นต้องใช้อย่างต่อเนื่องในระยะยาว ไม่ควรหยุดด้วยตนเอง
การรักษาด้วยการแลกเปลี่ยนพลาสมา อาจไม่ครอบคลุมในประกันสำหรับโรคประสาทตาอักเสบ ดังนั้นควรตรวจสอบกับสถานพยาบาลล่วงหน้า
Q
จะทำอย่างไรถ้าการรักษาด้วยสเตียรอยด์พัลส์ไม่ได้ผล?
A
การรักษาด้วยการแลกเปลี่ยนพลาสมา เป็นทางเลือกถัดไป โดยเลือกจาก 3 วิธี ได้แก่ การแลกเปลี่ยนพลาสมา อย่างง่าย การแลกเปลี่ยนพลาสมา ด้วยการกรองแบบเยื่อคู่ และการดูดซับภูมิคุ้มกัน การแลกเปลี่ยนพลาสมา อย่างง่ายถือว่ามีประสิทธิภาพสูงสุด แต่ก็มีภาระต่อร่างกายมากที่สุด ทำ 5-6 ครั้งต่อคอร์ส หลังการรักษาต้องเข้ารับการดูแลในโรงพยาบาล
NMOSD โดยพื้นฐานแล้วเป็นโรคของแอสโตรไซต์ (astrocytopathy) กลไกการเกิดโรคมีดังนี้
การสร้างแอนติบอดีและการผ่านด่านกั้นเลือด-สมอง (BBB)
ในส่วนปลาย เซลล์บีจะแยกตัวเป็นพลาสมาบลาสต์ที่หลั่ง AQP4-IgG IL-6 จะส่งเสริมการแยกตัวนี้และเพิ่มการซึมผ่านของ BBB บริเวณ area postrema เป็นบริเวณที่ไม่มี BBB และอาจเป็นเส้นทางที่ AQP4-IgG เข้าสู่ระบบประสาทส่วนกลาง
สายโซ่ของความเสียหายของแอสโตรไซต์
AQP4-IgG จับกับช่องน้ำ AQP4 ที่แสดงออกอย่างหนาแน่นบนส่วนปลายของแอสโตรไซต์ ทำให้เกิดความเสียหายของแอสโตรไซต์ผ่านวิถีทางดังต่อไปนี้
การกระตุ้นวิถีคลาสสิกของคอมพลีเมนต์ : ส่วน Fc ของ AQP4-IgG กระตุ้นคอมพลีเมนต์ สร้างเมมเบรนแอทแทคคอมเพล็กซ์ (MAC) และทำลายแอสโตรไซต์โดยตรงADCC (antibody-dependent cell-mediated cytotoxicity) : NK cells และนิวโทรฟิลทำลายแอสโตรไซต์ผ่าน Fcγ receptorการปล่อย C5a anaphylatoxin : กระตุ้นการเคลื่อนย้ายของ granulocyte (นิวโทรฟิลและอีโอซิโนฟิล) ทำให้เกิดความเสียหายของแอกซอนและการทำลายปลอกไมอีลินทุติยภูมิ1)
ความแตกต่างทางพยาธิวิทยาจาก MS
ใน MS การทำลายปลอกไมอีลินส่วนใหญ่เกิดในเนื้อขาวโดยมี CD8+ T cell เป็นหลัก แต่ใน NMOSD มีการเกี่ยวข้องของ CD4+ T cell มากกว่า และเกิดรอยโรคแบบเนื้อตายที่กระจายทั้งเนื้อเทาและเนื้อขาว
เหตุผลของการกระจายตัว
ช่อง AQP4 มีการกระจายตัวมากในเส้นประสาทตา บริเวณ postrema และไขสันหลัง ทำให้บริเวณเหล่านี้ถูกกำหนดเป้าหมายแบบเลือกสรร
ตัวบ่งชี้ทางชีวภาพ
GFA P ในซีรัม : สะท้อนความเสียหายของแอสโทรไซต์ และมีค่าสูงในช่วงที่มีอาการกำเริบนิวโรฟิลาเมนต์สายเบา (NfL) ในซีรัม : สะท้อนความเสียหายของแอกซอน และสัมพันธ์กับความรุนแรงของอาการกำเริบ1)
ไซโตไคน์ที่เกี่ยวข้อง ได้แก่ IL-6, IL-10, IL-17a, G-CSF, TNF -α และ BAFF/APRIL ตามรายงาน
เนื้อหาต่อไปนี้อยู่ในขั้นตอนการวิจัยหรือการทดลองทางคลินิก และไม่ใช่การรักษามาตรฐานที่สามารถรับได้ในโรงพยาบาลทั่วไป เป็นข้อมูลอ้างอิงสำหรับผู้เชี่ยวชาญเกี่ยวกับการพัฒนาทางการแพทย์ในอนาคต
ประมาณ 3-5% ของ NMOSD คาดว่าเป็นพารานีโอพลาสติก กรณีที่เกี่ยวข้องกับเทราโทมาของรังไข่ได้รับการศึกษาอย่างละเอียดเป็นพิเศษ
Ikeguchi และคณะ (2021) ได้ดำเนินการทบทวนผู้ป่วย NMOSD ที่มี AQP4+ ร่วมกับ teratoma รังไข่จำนวน 6 ราย2) ผู้ป่วยทั้งหมดเป็นเพศหญิง อายุเฉลี่ยเมื่อเริ่มป่วย 32.7 ปี (ช่วง 15-50 ปี) ในจำนวน 6 รายนี้ 83% (5/6 ราย) มีอาการคลื่นไส้อาเจียน 83% มี oligoclonal band ในน้ำไขสันหลังเป็นบวก และ 83% พบรอยโรคที่ dorsal brainstem การวิเคราะห์ทางพยาธิวิทยาพบว่าเนื้อเยื่อประสาทที่ติด GFA P ภายในเนื้องอกมี AQP4 immunoreactivity และมีการแทรกซึมของลิมโฟไซต์ ซึ่งบ่งชี้ว่าการนำเสนอ AQP4 antigen ภายในเนื้องอกกระตุ้นให้เกิด autoimmune response หลังการผ่าตัดเนื้องอกออก 60% (3/5 ราย) มีการเปลี่ยนเป็นลบของ AQP4-IgG
Ding และคณะ (2021) ได้ดำเนินการทบทวนผู้ป่วย NMOSD ที่เกี่ยวข้องกับเนื้องอกจำนวน 43 ราย3) ร้อยละ 88.4 เป็นผู้หญิง และมะเร็งเต้านมกับมะเร็งปอดเป็นชนิดเนื้องอกที่พบบ่อยที่สุด โดยเฉพาะในผู้ป่วย NMOSD ที่มีอายุ 50 ปีขึ้นไป เน้นย้ำถึงความสำคัญของการตรวจคัดกรองเนื้องอก
แนะนำให้ตรวจคัดกรองเนื้องอก รวมถึงเทอราโทมา แม้ในผู้ป่วยอายุน้อยก็ตาม
สำหรับ NMOSD ที่ดื้อต่อการรักษา ซึ่งไม่ตอบสนองต่อสเตียรอยด์ การแลกเปลี่ยนพลาสมา หรือริทูซิแมบ มีรายงานประสิทธิผลของการบำบัดด้วยการดูดซับอิมมูโนโกลบูลินชนิด Protein-A (IA)
Fan และคณะ (2024) รายงานผู้ป่วยหญิงอายุ 35 ปีที่มี NMOSD ร่วมกับ Sjögren’s syndrome ที่ดื้อต่อการรักษาด้วย steroid pulse และ IVI G โดยทำการรักษาด้วย Protein-A immunoadsorption จำนวน 3 ครั้ง4) ภายใน 1 สัปดาห์ อาการทางสายตา อาการอ่อนแรงครึ่งล่าง และการรับรู้ความรู้สึกผิดปกติดีขึ้นอย่างชัดเจน และพบว่าระดับ AQP4-IgG, IgA, IgG และ IgM ลดลงอย่างรวดเร็ว ในการติดตามผลเป็นเวลา 4 ปี ไม่พบการกลับเป็นซ้ำหรือการดำเนินโรค
ปัจจุบันเริ่มมีความชัดเจนเกี่ยวกับอุบัติการณ์ของโรคร่วม autoimmune ต่างๆ ใน NMOSD
Zhu และคณะ (2025) รายงานผู้ป่วยเด็กหญิงอายุ 14 ปีที่เริ่มมี NMOSD เมื่ออายุ 11 ปี5) ผลตรวจ AQP4-IgG เป็นบวก และในระหว่างการดำเนินโรคพบว่ามี primary Sjögren’s syndrome ร่วมด้วย ผู้ป่วยได้รับการรักษาด้วย methylprednisolone, IVI G, mycophenolate mofetil (MMF) และเปลี่ยนเป็น tacrolimus ทำให้โรคสงบ มีรายงานว่าผู้ใหญ่ที่เป็น NMOSD ร้อยละ 20-30 มีโรคร่วม autoimmune แต่รายงานนี้แสดงให้เห็นว่าการเกิดร่วมกันก็พบได้ในผู้ป่วยเด็กเช่นกัน5)
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, et al. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD : A case report and review. Brain Behav. 2021;11(9):e2282.
Fan W, Chen X, Xiao P, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD : a case report and literature review. Front Immunol. 2024;15:1429405.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.