Nöromiyelitis optika spektrum bozukluğu (NMOSB), merkezi sinir sistemini etkileyen inflamatuvar, antikor aracılı, otoimmün bir hastalıktır. Eskiden “Devic hastalığı” olarak da adlandırılan bu hastalık, uzun yıllar multipl sklerozun (MS) bir alt tipi olarak kabul edilmiştir. Ancak 2004 yılında aquaporin-4’e (AQP4) karşı otoantikorların (AQP4-IgG) keşfedilmesiyle bağımsız bir hastalık birimi olarak tanımlanmıştır.
Tarihsel arka plan olarak, 1870 yılında Sir Thomas Clifford Allbutt, miyelit ve optik nöropati arasındaki ilişkiyi ilk kez tanımlamıştır. Daha sonraki araştırmalar, bu durumun MS’ten farklı bir hastalık olduğunu ortaya koymuş ve günümüzde “NMOSB” kapsayıcı kavramı altında ele alınmaktadır.
Epidemiyoloji aşağıdaki gibidir:
İnsidans: AQP4+ NMOSB’nin tahmini yıllık insidansı 0,4-7,3/milyon kişidir1)
Cinsiyet farkı: Kadın/erkek oranı yaklaşık 1:9 olup kadınlarda belirgin şekilde daha sıktır
Başlangıç yaşı: Genellikle orta yaşta (40-60 yaş) ortaya çıkar. Japonya’da pik yaş 30’lu yaşların sonu ile 40’lı yaşların başıdır
Irk: Afrika kökenli ve Asya kökenli popülasyonlarda daha sık görülme eğilimindedir1)
Gebelikle ilişkisi: Kadınların yaklaşık %20-47’si gebelik sırasında veya doğum/düşükten sonraki bir yıl içinde ilk atağı geçirir.
QNMOSD ve multipl skleroz (MS) arasındaki fark nedir?
A
NMOSD, astrositlerdeki AQP4 su kanallarını hedef alan antikor aracılı bir hastalıktır ve patogenez, tedavi ve prognoz açısından MS’ten temel olarak farklıdır. NMOSD’de LETM ve şiddetli optik nörit tipiktir ve MS’te etkili olan interferon beta gibi hastalık modifiye edici ilaçlar NMOSD’de atakları tetikleyebilir, bu da önemli bir farktır.
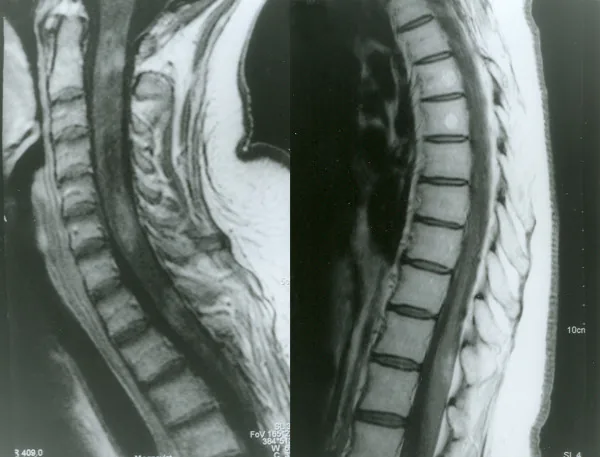
Petelin Gadze Z, et al. Patient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case report. BMC Neurol. 2009. Figure 2. PMCID: PMC2773232. License: CC BY.
Nisan 2007’de çekilen omurilik gadolinyum kontrastlı T1 ağırlıklı MRG görüntüsü olup, C2, C4-C5 ve Th1 seviyelerinde heterojen halkasal kontrast tutulumu izlenmektedir. Metnin “2. Başlıca belirtiler ve klinik bulgular” bölümünde ele alınan miyelite karşılık gelir.
NMOSD’nin belirtileri etkilenen bölgeye göre çeşitlilik gösterir.
Ani görme kaybı: Başlıca semptomlardan biri. Steroid tedavisine direnç göstermesi karakteristiktir.
Göz ağrısı: Optik nörite eşlik eder ve vakaların yaklaşık yarısında görülür.
Renk görme bozukluğu: Kırmızı renk doygunluğunda azalma tipiktir.
Görme alanı defekti: Santral skotomla sınırlı kalmayıp, horizontal hemianopsi, bitemporal hemianopsi ve homonim hemianopsi de görülebilir. Bunun nedeni lezyonun kiazma ve optik traktusa kadar uzanmasıdır.
Duyu kusuru ve parapleji: Miyelite bağlı motor ve duyu bozuklukları.
Mesane ve rektum disfonksiyonu: Miyelite eşlik eden otonom sinir sistemi bozukluğu.
Tedaviye dirençli hıçkırık, bulantı ve kusma: Area postrema lezyonuna bağlı karakteristik semptomlar.
Göz hareket bozukluğu: Beyin sapı lezyonuna bağlı.
Aşırı uyku hali (narkolepsi benzeri): Diensefalon/hipotalamus lezyonuna bağlı
Hastalığın başlangıcında grip benzeri semptomlar (ateş, kas ağrısı, baş ağrısı) görülebilir.
Optik disk ödemi: Akut dönemde görülür, daha sonra optik atrofiye ilerler.
RAPD (Rölatif afferent pupil defekti): Tek veya iki taraflı olabilir.
Bilateral eşzamanlı: NMOSD’de optik nörit %17-82 oranında iki taraflı eşzamanlıdır. MS’ten önemli fark.
Ciddi görme kaybı: AQP4+ NMOSD’de en düşük görme keskinliği ortancası el hareketi (HM) düzeyindedir. İyileşme sonrası ortanca parmak sayma düzeyinde kalır ve %60-69’u en az bir gözde 20/200 veya altında kalıcı görme bozukluğu bırakır.
Miyelit
LETM (Uzun Boyuna Enine Miyelit): Üç veya daha fazla vertebrayı kapsayan sürekli lezyon. AQP4+ NMOSD’li hastaların yaklaşık %85’i akut miyelit sırasında bunu gösterir.
Komple Medüller Sendrom: Motor, duyusal ve otonomik olmak üzere üç yolu da etkiler.
Ciddi Fonksiyonel Bozukluk: Hastaların %30’undan fazlası atak sırasında tekerlekli sandalyeye bağımlı hale gelir. AQP4+ NMOSD’li hastaların %37-44’ü sonunda yürüme yardımcısına ihtiyaç duyar.
Area Postrema Sendromu
İnatçı Hıçkırık: Günler ila haftalar sürer ve standart antiemetiklere yanıt vermez.
Bulantı ve Kusma: Area postrema kan-beyin bariyerinden yoksun olduğu için AQP4-IgG’nin doğrudan ulaşması kolaydır.
NMOSD Tanısında Temel Bulgu: Açıklanamayan inatçı hıçkırık, NMOSD’nin aktif olarak düşünülmesi için bir fırsattır.
QNMOSD'ye bağlı optik nörit, MS ile ilişkili olandan nasıl farklıdır?
A
NMOSD’ye bağlı optik nörit daha şiddetli, iki taraflı ve tekrarlayıcı olup görme prognozu kötüdür. AQP4-pozitif NMOSD’de hastaların %60-69’unda en az bir gözde 20/200 veya daha kötü kalıcı görme kaybı geliştiği bildirilmiştir. Ayrıca kiazmayı tutmaya yatkın olup bitemporal hemianopsi gibi çeşitli görme alanı defektlerine yol açması da MS’den farklıdır.
Spinal MRI: LETM en karakteristik bulgudur. Santral gri cevher baskındır. Spinal kord şişliği, T1 hipointensitesi ve Gd kontrastlanması eşlik eder. AQP4+ NMOSD’li hastaların yaklaşık %85’i akut miyelit sırasında LETM gösterir1)
Optik sinir MRI: Yağ baskılamalı görüntüleme zorunludur. Bilateral ve uzun segment inflamasyon (%50’den fazla) karakteristiktir. Posterior kısım ve kiazma tutulumu AQP4+ NMOSD için tipiktir1)
Beyin MRG: Area postrema lezyonları, 4. ventrikül çevresi beyin sapı lezyonları, hipotalamus/3. ventrikül çevresi lezyonları, yaygın beyaz cevher lezyonları vb. görülür.
NMOSD’nin özellikleri: MS’ten farklı olarak, asemptomatik yeni T2 lezyonları nadirdir (%3-13). Sürveyans MRG genellikle gerekli değildir1)
Yaşlılarda iskemik optik nöropati, servikal spondilotik miyelopati, spinal enfarktüs ve primer CNS lenfoması ile ayırıcı tanı önemlidir.
QAQP4 antikoru negatif olsa bile NMOSD tanısı konulabilir mi?
A
Evet. AQP4-IgG negatif veya test edilmemiş olsa bile, iki veya daha fazla ana klinik özellik karşılanırsa, ek MRI gereksinimleri karşılanırsa ve diğer hastalıklar dışlanırsa NMOSD tanısı konulabilir. Ayrıca, AQP4-IgG negatif vakaların yaklaşık %30’unda MOG-IgG pozitiftir ve her iki antikorun ölçümü önerilir. AQP4-IgG negatif vakaların %1’inden azının daha sonra serokonversiyon geçirdiği bildirilmiştir.
Metilprednizolon 1000 mg/gün intravenöz infüzyon 3 gün boyunca uygulanır.
Görme düzelmezse, 3-4 gün ara ile ikinci bir kür uygulanması düşünülür.
NMOSD’ye bağlı optik nörit, steroidlere karşı yüksek direnç gösterdiğinden, yanıt yetersizse erken dönemde bir sonraki tedavi düşünülmelidir.
İkinci seçenek: Plazma değişimi tedavisi
Steroid pulse tedavisine yanıt alınamadığında uygulanır. Aşağıdaki yöntemler seçenek olarak değerlendirilir.
Basit plazma değişimi (PE): Etkisi en yüksektir ancak vücuda verdiği hasar da en fazladır.
Çift membran filtrasyon plazma değişimi (DFPP)
İmmün adsorpsiyon tedavisi (IA): Antikorların seçici olarak uzaklaştırılmasını sağlar.
Etkinlik sırası basit plazma değişimi > çift membran filtrasyon > immün adsorpsiyon şeklindedir. Bir kürde 5-6 seans uygulanır ve tedavi sonrası vücut IgG düzeyi düzelene kadar hastanede yatış gerekir. Optik nörit için bu tedavilerin sigorta kapsamı dışında kalabileceği unutulmamalı ve hastaya bu konuda bilgi verilmelidir.
AQP4+NMOSD’de ilk ataktan sonra idame tedavisine erken başlanması gerektiği düşünülmektedir1). Japonya’da plazma değişiminden sonra prednizolon 5-10 mg/gün + azatioprin 50-100 mg/gün’e geçiş yaygındır.
Kanıt düzeyi yüksek biyolojik ajanlar şunlardır:
Kompleman inhibitörleri
Ekulizumab: 900 mg IV haftada bir ×4 kez → 1.200 mg 2 haftada bir idame dozu1)
Ravulizumab: Vücut ağırlığına dayalı yükleme dozu (2.400-3.000 mg) → 15. günden itibaren 3.000-3.600 mg 8 haftada bir1)
B hücre yok edici tedaviler
Rituksimab: 375 mg/m² IV haftada bir ×4 kez veya 1.000 mg ×2 kez (2 hafta arayla) → 6 ayda bir 1.000 mg ×2 kez1)
İnebilizumab: 300 mg IV 15 günde bir 2 kez → 6 ayda bir1)
Plazma değişimi tedavisi bir sonraki seçenektir. Basit plazma değişimi, çift filtrasyon plazma değişimi ve immün adsorpsiyon olmak üzere üç tür arasından seçim yapılır. Basit plazma değişimi en etkili olarak kabul edilir ancak vücut üzerinde daha fazla yük oluşturur. Bir kürde 5-6 seans uygulanır ve tedavi sonrası hastanede yatış gerekir.
NMOSD temelde bir astrosit hastalığıdır (astrositopati). Patogenez aşağıdaki gibidir.
Antikor üretimi ve kan-beyin bariyerini (KBB) geçiş
Periferde B hücreleri, AQP4-IgG salgılayan plazmablastlara farklılaşır. IL-6 bu farklılaşmayı teşvik eder ve KBB geçirgenliğini artırır. Area postrema, KBB’si olmayan bir bölgedir ve AQP4-IgG’nin MSS’ye giriş yolu olabilir.
Astrosit hasar zinciri
Astrosit ayak uçlarında yoğun olarak eksprese edilen AQP4 su kanallarına AQP4-IgG bağlanır ve aşağıdaki yollarla astrosit hasarı oluşur.
Kompleman klasik yolunun aktivasyonu: AQP4-IgG’nin Fc kısmı komplemanı aktive eder, membran atak kompleksi (MAC) oluşturarak astrositlere doğrudan zarar verir
ADCC (antikor bağımlı hücresel sitotoksisite): NK hücreleri ve nötrofiller, Fcγ reseptörleri aracılığıyla astrositlere zarar verir
C5a anafilatoksin salınımı: Granülositleri (nötrofil, eozinofil) harekete geçirerek sekonder akson hasarı ve demiyelinizasyona neden olur1)
MS’te esas olarak CD8+ T hücrelerinin merkezi rol oynadığı beyaz cevher ağırlıklı demiyelinizasyon görülürken, NMOSD’de CD4+ T hücrelerinin katılımı daha fazladır ve hem gri cevher hem de beyaz cevheri içeren nekrotik lezyonlar oluşur.
Dağılımın nedeni
AQP4 kanalları optik sinir, area postrema ve omurilikte yoğun olarak bulunur; bu bölgeler seçici olarak hedef alınır.
Biyobelirteçler
Serum GFAP: Astrosit hasarını yansıtır, atak sırasında yüksek değerlere ulaşır
Serum nörofilaman hafif zinciri (NfL): Akson hasarını yansıtır, atak şiddeti ile korelasyon gösterir1)
İlgili sitokinler olarak IL-6, IL-10, IL-17a, G-CSF, TNF-α ve BAFF/APRIL rapor edilmiştir.
7. Güncel Araştırmalar ve Gelecek Perspektifleri (Araştırma Aşamasındaki Raporlar)
NMOSD vakalarının %3-5’inin paraneoplastik olduğu tahmin edilmektedir. Over teratomu ile ilişkili vakalar özellikle detaylı olarak araştırılmıştır.
Ikeguchi ve ark. (2021), over teratomu ile ilişkili AQP4+ NMOSD’nin 6 vakalık bir incelemesini gerçekleştirmiştir2). Tüm vakalar kadındı, ortalama başlangıç yaşı 32,7 (15-50). Altı vakanın %83’ünde (5/6) bulantı ve kusma görüldü, %83’ünde BOS oligoklonal bandı pozitifti ve %83’ünde dorsal beyin sapı lezyonları mevcuttu. Patolojik incelemede, tümör içindeki GFAP-pozitif sinir dokusunda AQP4 immünreaktivitesi ve lenfosit infiltrasyonu doğrulandı; bu, tümör içinde AQP4 antijen sunumunun otoimmün yanıtı tetiklediği mekanizmayı düşündürmektedir. Tümör çıkarılmasından sonra vakaların %60’ında (3/5) AQP4-IgG negatifleşti.
Ding ve ark. (2021), 43 paraneoplastik NMOSD vakasını incelemiştir3). %88,4’ü kadındı ve en sık görülen tümör tipleri meme kanseri ve akciğer kanseriydi. Özellikle 50 yaş üstü NMOSD hastalarında tümör taramasının önemi vurgulanmaktadır.
Genç hastalarda bile teratom dahil tümör taraması önerilmektedir.
Fan ve ark. (2024), steroid pulse ve IVIG’ye yanıt vermeyen refrakter Sjögren sendromu ile ilişkili NMOSD’li 35 yaşında bir kadın hastaya 3 seans Protein-A immünadsorpsiyon tedavisi uygulamıştır4). Bir hafta içinde görme bozukluğu, parapleji ve propriyoseptif duyu kaybı belirgin şekilde düzelmiş ve AQP4-IgG, IgA, IgG, IgM’de hızlı düşüş gözlenmiştir. Dört yıllık takipte nüks veya ilerleme saptanmamıştır.
Otoimmün hastalıklarla birliktelik gösteren vakaların özellikleri
NMOSD’ye çeşitli otoimmün hastalıkların eşlik etme durumu giderek daha iyi anlaşılmaktadır.
Zhu ve ark. (2025), 11 yaşında NMOSD gelişen 14 yaşında bir kız çocuğu vakasını bildirmiştir5). AQP4-IgG pozitifti ve takipte primer Sjögren sendromu birlikteliği doğrulandı. Metilprednizolon, IVIG ve mikofenolat mofetil (MMF) → takrolimus değişimi ile remisyon sağlanmıştır. Erişkin NMOSD vakalarının %20-30’unda otoimmün hastalık birlikteliği bildirilirken, çocuk vakalarda da birlikteliğin olabileceği gösterilmiştir5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, et al. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain Behav. 2021;11(9):e2282.
Fan W, Chen X, Xiao P, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Front Immunol. 2024;15:1429405.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.