O transtorno do espectro da neuromielite óptica (NMOSD) é uma doença inflamatória, autoimune e mediada por anticorpos que afeta o sistema nervoso central. Anteriormente chamada de “doença de Devic”, foi por muitos anos considerada uma variante da esclerose múltipla (EM). No entanto, a descoberta em 2004 de autoanticorpos contra a aquaporina-4 (AQP4-IgG) estabeleceu-a como uma entidade clínica distinta.
Contexto histórico: Em 1870, Sir Thomas Clifford Allbutt descreveu pela primeira vez a associação entre mielite e distúrbios do nervo óptico. Estudos posteriores reconheceram-na como uma doença diferente da EM, e atualmente é compreendida sob o conceito abrangente de “NMOSD”.
Epidemiologia é a seguinte:
Incidência: A incidência anual estimada de NMOSD AQP4+ é de 0,4 a 7,3 por milhão de pessoas1)
Sexo: A proporção entre homens e mulheres é de aproximadamente 1:9, com predomínio acentuado no sexo feminino
Idade de início: Ocorre principalmente na meia-idade (40 a 60 anos). No Japão, o pico ocorre no final dos 30 anos ao início dos 40 anos
Raça: Tendência a ser mais comum em populações africanas e asiáticas1)
Relação com a gravidez: cerca de 20 a 47% das mulheres apresentam o primeiro episódio durante a gravidez ou dentro de um ano após o parto ou aborto
QQual é a diferença entre NMOSD e esclerose múltipla (EM)?
A
A NMOSD é uma doença mediada por anticorpos direcionados aos canais de água AQP4 nos astrócitos, diferindo fundamentalmente da EM em patologia, tratamento e prognóstico. Na NMOSD, são típicas a LETM e a neurite óptica grave, e uma diferença importante é que medicamentos modificadores da doença eficazes na EM, como o interferon beta, podem desencadear recaídas na NMOSD.
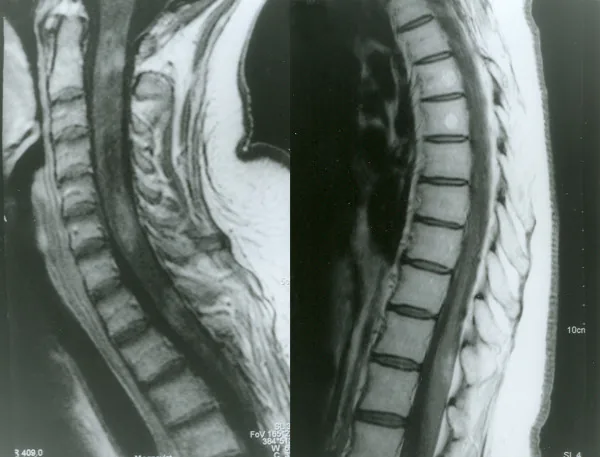
Ressonância magnética com contraste da medula espinhal na neuromielite óptica
Petelin Gadze Z, et al. Patient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case report. BMC Neurol. 2009. Figure 2. PMCID: PMC2773232. License: CC BY.
Imagem de ressonância magnética ponderada em T1 com gadolínio da medula espinhal, adquirida em abril de 2007, mostrando realce anelar heterogêneo nos níveis C2, C4-C5 e Th1, correspondente à mielite discutida na seção “2. Principais sintomas e achados clínicos”.
Os sintomas da NMOSD variam conforme a região afetada.
Perda súbita da visão: um dos principais sintomas. Caracteriza-se pela resistência ao tratamento com corticosteroides
Dor ocular: associada à neurite óptica, presente em cerca de metade dos casos
Alteração da visão de cores: tipicamente, diminuição da saturação do vermelho
Defeito de campo visual: não se limita a escotoma central; pode ocorrer hemianopsia horizontal, hemianopsia bitemporal ou hemianopsia homônima, devido ao envolvimento do quiasma óptico e do trato óptico
Alterações sensitivas e paraplegia: déficits motores e sensitivos devido à mielite
Disfunção vesical e retal: distúrbios autonômicos associados à mielite
Soluços intratáveis e náuseas/vômitos: sintomas característicos devido à lesão da área postrema
Alteração dos movimentos oculares: devido a lesão do tronco encefálico
Hipersônia (narcolepsia-like): devido a lesão diencefálica/hipotalâmica
No início da doença, podem ocorrer sintomas gripais (febre, mialgia, cefaleia).
Bilateral simultânea: a neurite óptica na NMOSD é bilateral simultânea em 17-82% dos casos. Diferença importante em relação à EM.
Perda visual grave: na NMOSD AQP4+, a mediana da pior acuidade visual é de movimento de mãos (MM). Após recuperação, a mediana é de conta dedos (CD), e 60-69% dos pacientes mantêm deficiência visual permanente de ≤20/200 em pelo menos um olho.
Mielite
LETM (Mielite Transversa Longitudinalmente Extensa): Lesão contínua abrangendo 3 ou mais corpos vertebrais. Cerca de 85% dos casos de NMOSD AQP4+ apresentam isso durante a mielite aguda.
Síndrome Medular Completa: Envolvimento de todas as três vias: motora, sensitiva e autonômica.
Disfunção Grave: Mais de 30% dos pacientes tornam-se dependentes de cadeira de rodas no pior momento do ataque. 37-44% dos pacientes com NMOSD AQP4+ eventualmente necessitam de auxílio para caminhar.
Síndrome da Área Postrema
Soluços Intratáveis: Persistem por dias a semanas e não respondem a antieméticos convencionais.
Náuseas e Vômitos: Devido à ausência de barreira hematoencefálica na área postrema, local onde a AQP4-IgG pode atingir diretamente.
Achado Central para Diagnóstico de NMOSD: Soluços intratáveis inexplicáveis são um motivo para suspeitar ativamente de NMOSD.
QComo a neurite óptica na NMOSD difere da relacionada à EM?
A
A neurite óptica na NMOSD é mais grave, bilateral e recorrente, com pior prognóstico visual. Na NMOSD AQP4+, estima-se que 60-69% dos pacientes apresentam deficiência visual permanente de 20/200 ou pior em pelo menos um olho. Além disso, o envolvimento do quiasma óptico é mais comum, causando diversos defeitos de campo visual, como hemianopsia bitemporal, o que também a diferencia da EM.
RM da medula espinhal: LETM é a mais característica. Predomínio da substância cinzenta central. Acompanhada de edema medular, hipossinal em T1 e realce por Gd. Cerca de 85% dos AQP4+NMOSD apresentam LETM durante mielite aguda1)
RM do nervo óptico: Imagens com supressão de gordura são essenciais. Inflamação bilateral e de longo segmento (>50%) é característica. Envolvimento da porção posterior e do quiasma óptico é típico do AQP4+NMOSD1)
RM cerebral: lesões na área postrema, tronco encefálico periventricular do quarto ventrículo, hipotálamo/terceiro ventrículo e lesões extensas da substância branca
Características da NMOSD: ao contrário da EM, novas lesões T2 assintomáticas são raras (3–13%). A RM de vigilância geralmente não é necessária1)
Em idosos, é importante o diagnóstico diferencial com neuropatia óptica isquêmica, mielopatia cervical espondilótica, infarto medular e linfoma primário do SNC
QO diagnóstico de NMOSD pode ser feito mesmo com anticorpo AQP4 negativo?
A
Sim. Mesmo com AQP4-IgG negativo ou não testado, o diagnóstico de NMOSD pode ser feito se dois ou mais achados clínicos principais estiverem presentes, os critérios adicionais de RM forem atendidos e outras doenças forem excluídas. Além disso, cerca de 30% dos casos AQP4-IgG negativos são positivos para MOG-IgG, sendo recomendada a dosagem de ambos os anticorpos. Menos de 1% dos casos AQP4-IgG negativos soroconvertem posteriormente.
Metilprednisolona 1.000 mg/dia por via intravenosa por 3 dias
Se não houver melhora visual, considerar um novo ciclo após intervalo de 3 a 4 dias
A neurite óptica na NMOSD é altamente resistente a esteroides; portanto, se a resposta for insuficiente, considere o próximo tratamento precocemente.
Segunda linha: Plasmaférese
Realizada quando não há resposta ao pulso de esteroide. As seguintes opções estão disponíveis.
Plasmaférese simples (PE): Maior eficácia, mas também maior dano ao organismo.
Plasmaférese de dupla filtração (DFPP)
Imunoadsorção (IA): Permite remoção seletiva de anticorpos.
A ordem de eficácia é plasmaférese simples > dupla filtração > imunoadsorção. Realiza-se 5 a 6 sessões por ciclo, e após o tratamento é necessária internação até a recuperação dos níveis de IgG. Observe que pode não ser coberta pelo seguro para “neurite óptica”, sendo necessário explicar ao paciente.
Na NMOSD AQP4+, o tratamento de manutenção deve ser iniciado precocemente após o primeiro ataque 1). No Japão, após a plasmaférese, a transição para prednisolona 5–10 mg/dia + azatioprina 50–100 mg/dia é comum.
Agentes biológicos com alto nível de evidência são os seguintes:
Inibidores do complemento
Eculizumabe: 900 mg IV semanalmente × 4 → manutenção de 1.200 mg a cada 2 semanas 1)
Ravulizumabe: dose de ataque baseada no peso (2.400–3.000 mg) → a partir do dia 15, 3.000–3.600 mg a cada 8 semanas 1)
Terapia de depleção de células B
Rituximabe: 375 mg/m² IV semanalmente × 4, ou 1.000 mg × 2 (com intervalo de 2 semanas) → 1.000 mg × 2 a cada 6 meses 1)
Inebilizumabe: 300 mg IV a cada 15 dias × 2 → a cada 6 meses 1)
Inibidor do receptor de IL-6
Satralizumabe: 120 mg por via subcutânea a cada 4 semanas1)
QO que fazer se a pulsoterapia com corticoides não funcionar?
A
A plasmaférese é a próxima opção. Existem três tipos: plasmaférese simples, plasmaférese por dupla filtração e imunoadsorção. A plasmaférese simples é considerada a mais eficaz, mas também impõe maior carga ao organismo. São realizadas 5 a 6 sessões por ciclo, sendo necessária internação hospitalar após o tratamento.
A NMOSD é essencialmente uma astrocitopatia. O mecanismo da doença é o seguinte:
Produção de anticorpos e passagem pela barreira hematoencefálica (BHE)
Na periferia, os linfócitos B diferenciam-se em plasmablastos secretores de AQP4-IgG. A IL-6 promove essa diferenciação e aumenta a permeabilidade da BHE. A área postrema é uma região sem BHE, podendo servir como via de entrada da AQP4-IgG no SNC.
Cascata de lesão astrocitária
A AQP4-IgG liga-se aos canais de água AQP4, altamente expressos nos pés dos astrócitos, causando lesão astrocitária pelas seguintes vias:
Ativação da via clássica do complemento: a porção Fc da AQP4-IgG ativa o complemento, formando o complexo de ataque à membrana (MAC) que lesa diretamente os astrócitos
ADCC (citotoxicidade celular dependente de anticorpos): células NK e neutrófilos lesam os astrócitos via receptores Fcγ
Liberação de anafilatoxina C5a: recruta granulócitos (neutrófilos e eosinófilos), causando dano axonal secundário e desmielinização1)
Diferenças patológicas em relação à EM
Na EM, a desmielinização é predominantemente na substância branca, com papel central dos linfócitos T CD8+, enquanto na NMOSD há maior envolvimento de linfócitos T CD4+, formando lesões necróticas que afetam tanto a substância cinzenta quanto a branca.
Razão da distribuição
Os canais AQP4 são abundantemente distribuídos no nervo óptico, área postrema e medula espinhal, tornando essas regiões alvos seletivos.
Biomarcadores
GFAP sérico: reflete dano astrocitário, elevando-se durante as crises
Cadeia leve de neurofilamento sérico (NfL): reflete dano axonal e correlaciona-se com a gravidade das crises1)
As citocinas envolvidas incluem IL-6, IL-10, IL-17a, G-CSF, TNF-α e BAFF/APRIL.
7. Pesquisas recentes e perspectivas futuras (relatos em fase de pesquisa)
Estima-se que 3 a 5% dos casos de NMOSD sejam paraneoplásicos. Casos associados a teratoma ovariano têm sido particularmente estudados.
Ikeguchi et al. (2021) realizaram uma revisão de 6 casos de NMOSD AQP4+ associada a teratoma ovariano2). Todos os casos eram mulheres, com idade média de início de 32,7 anos (15 a 50 anos). Dos 6 casos, 83% (5/6) apresentaram náuseas e vômitos, 83% tiveram bandas oligoclonais positivas no LCR e 83% apresentaram lesões no tronco encefálico dorsal. A análise patológica confirmou imunorreatividade para AQP4 e infiltração linfocitária no tecido neural GFAP-positivo dentro do tumor, sugerindo que a apresentação do antígeno AQP4 no tumor desencadeia uma resposta autoimune. Após a ressecção tumoral, 60% (3/5) dos casos tiveram soroconversão negativa para AQP4-IgG.
Ding et al. (2021) realizaram uma revisão de 43 casos de NMOSD paraneoplásica 3). 88,4% eram mulheres, e câncer de mama e pulmão foram os tipos tumorais mais frequentes. A importância da triagem tumoral é enfatizada especialmente em pacientes com NMOSD com mais de 50 anos.
Recomenda-se a triagem de tumores, incluindo teratomas, mesmo em pacientes jovens.
A eficácia da terapia de imunoadsorção com proteína A (IA) foi relatada para NMOSD refratária que não responde a esteroides, plasmaférese e rituximabe.
Fan et al. (2024) realizaram 3 sessões de imunoadsorção com Proteína-A em uma mulher de 35 anos com NMOSD associada à síndrome de Sjögren refratária, que não respondia a pulsoterapia com esteroides e IVIG4). Dentro de uma semana, houve melhora acentuada da deficiência visual, paraplegia e distúrbios de sensibilidade proprioceptiva, com rápida redução de AQP4-IgG, IgA, IgG e IgM. Nenhuma recidiva ou progressão foi observada durante 4 anos de acompanhamento.
Características dos casos de comorbidade com doenças autoimunes
A realidade da coexistência de várias doenças autoimunes com NMOSD está se tornando mais clara.
Zhu et al. (2025) relataram o caso de uma menina de 14 anos que desenvolveu NMOSD aos 11 anos5). AQP4-IgG positiva, e durante o curso foi confirmada a presença de síndrome de Sjögren primária. A remissão foi mantida com metilprednisolona, IVIG e micofenolato de mofetil (MMF), posteriormente alterado para tacrolimus. Em adultos com NMOSD, 20-30% apresentam comorbidade com doenças autoimunes, mas este caso demonstra que essa associação também ocorre em crianças5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, et al. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain Behav. 2021;11(9):e2282.
Fan W, Chen X, Xiao P, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Front Immunol. 2024;15:1429405.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.