Neuromyelitis optica spectrum disorder (NMOSD) is an inflammatory, antibody-mediated, autoimmune disease that affects the central nervous system. Previously called “Devic’s disease,” it was long considered a subtype of multiple sclerosis (MS). However, the discovery of autoantibodies against aquaporin-4 (AQP4-IgG) in 2004 established it as an independent disease entity.
Historical background: In 1870, Sir Thomas Clifford Allbutt first described the association between myelitis and optic neuropathy. Subsequent research recognized it as a disease distinct from MS, and it is now understood under the comprehensive concept of “NMOSD.”
Epidemiology is as follows:
Incidence: The estimated annual incidence of AQP4+ NMOSD is 0.4–7.3 per million people1)
Sex ratio: The male-to-female ratio is approximately 1:9, with a marked predominance in women
Age of onset: Mainly occurs in middle age (40–60 years). In Japan, the peak is in the late 30s to early 40s
Race: Tends to be more common in African and Asian populations1)
Association with pregnancy: Approximately 20–47% of women experience their first onset during pregnancy or within one year after childbirth or miscarriage.
QHow is NMOSD different from multiple sclerosis (MS)?
A
NMOSD is an antibody-mediated disease targeting the AQP4 water channel on astrocytes, and its pathology, treatment, and prognosis are fundamentally different from MS. In NMOSD, LETM and severe optic neuritis are typical, and an important difference is that disease-modifying drugs effective for MS, such as interferon beta, may trigger relapses in NMOSD.
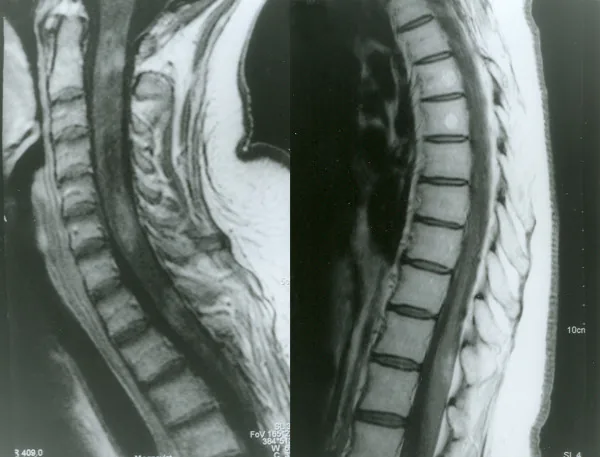
Contrast-enhanced MRI of the spinal cord in neuromyelitis optica
Petelin Gadze Z, et al. Patient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case report. BMC Neurol. 2009. Figure 2. PMCID: PMC2773232. License: CC BY.
Gadolinium-enhanced T1-weighted MRI of the spinal cord taken in April 2007, showing heterogeneous ring enhancement at C2, C4-C5, and Th1 levels, corresponding to myelitis discussed in section “2. Main Symptoms and Clinical Findings”.
The symptoms of NMOSD vary depending on the affected area.
Rapid vision loss: One of the main symptoms. It is characterized by resistance to steroid therapy.
Eye pain: Associated with optic neuritis, occurring in about half of cases.
Color vision abnormality: Typically a decrease in red saturation.
Visual field defects: Not limited to central scotoma; may also cause horizontal hemianopia, bitemporal hemianopia, or homonymous hemianopia. This is because lesions extend to the optic chiasm and optic tract.
Sensory disturbance and paraplegia: Motor and sensory deficits due to myelitis
Bladder and rectal dysfunction: Autonomic neuropathy associated with myelitis
Intractable hiccups and nausea/vomiting: Characteristic symptoms due to area postrema lesions
Oculomotor disturbance: Due to brainstem lesions
Hypersomnia (narcolepsy-like): Due to diencephalic/hypothalamic lesions
In the early stage, influenza-like symptoms (fever, myalgia, headache) may occur.
Bilateral simultaneous onset: Optic neuritis in NMOSD is bilateral in 17–82% of cases. An important distinction from MS.
Severe vision loss: In AQP4+ NMOSD, the median nadir visual acuity is hand motion (HM) level. After recovery, the median visual acuity remains counting fingers, and 60–69% have permanent visual impairment of 20/200 or worse in at least one eye.
Myelitis
LETM (Longitudinally Extensive Transverse Myelitis): Continuous lesion spanning 3 or more vertebral segments. Approximately 85% of AQP4+ NMOSD cases show this during acute myelitis.
Complete spinal cord syndrome: Involves all three pathways—motor, sensory, and autonomic.
Severe functional impairment: Over 30% of patients are wheelchair-dependent at the nadir of an attack. 37–44% of AQP4+ NMOSD patients eventually require walking aids.
Area Postrema Syndrome
Intractable hiccups: Persist for days to weeks and do not respond to standard antiemetics.
Nausea and vomiting: This occurs because the area postrema lacks a blood-brain barrier, making it a site where AQP4-IgG can directly reach.
Core diagnostic findings of NMOSD: Unexplained intractable hiccups are a trigger to actively suspect NMOSD.
QHow does optic neuritis in NMOSD differ from that in MS?
A
Optic neuritis in NMOSD is more severe, bilateral, and recurrent, with poor visual prognosis. In AQP4+ NMOSD, 60–69% of patients are reported to have permanent visual impairment of 20/200 or worse in at least one eye. Additionally, it tends to involve the optic chiasm, leading to various visual field defects such as bitemporal hemianopia, which is a difference from MS.
Spinal cord MRI: LETM is most characteristic. Central gray matter predominance. Accompanied by spinal cord swelling, T1 hypointensity, and Gd enhancement. Approximately 85% of AQP4+ NMOSD present with LETM during acute myelitis 1)
Optic nerve MRI: Fat-suppressed imaging is essential. Bilateral, long-segment inflammation (≥50%) is characteristic. Involvement of the posterior portion and optic chiasm is characteristic of AQP4+ NMOSD1)
Brain MRI: Findings include area postrema lesions, fourth ventricle periventricular brainstem lesions, hypothalamic/third ventricle periventricular lesions, and extensive white matter lesions
Characteristics of NMOSD: Unlike MS, asymptomatic new T2 lesions are rare (3–13%). Surveillance MRI is usually unnecessary 1)
In elderly patients, differentiation from ischemic optic neuropathy, cervical spondylotic myelopathy, spinal cord infarction, and primary CNS lymphoma is important
QCan NMOSD be diagnosed even if AQP4 antibody is negative?
A
Even if AQP4-IgG is negative or untested, NMOSD can be diagnosed if two or more major clinical features are met, additional MRI requirements are satisfied, and other diseases are excluded. Additionally, about 30% of AQP4-IgG-negative cases are positive for MOG-IgG, and measurement of both antibodies is recommended. Less than 1% of AQP4-IgG-negative cases later seroconvert.
Intravenous methylprednisolone 1,000 mg/day for 3 days
If visual improvement is insufficient, consider repeating one course after an interval of 3–4 days.
Optic neuritis in NMOSD is highly resistant to steroids, so if response is insufficient, consider the next treatment early.
Second-line: Plasma exchange therapy
Performed when there is no response to steroid pulse therapy. The following methods are options.
Simple plasma exchange (PE): Most effective but also causes the greatest damage to the body.
Double filtration plasmapheresis (DFPP)
Immunoadsorption therapy (IA): Allows selective removal of antibodies.
The order of effectiveness is simple plasma exchange > double filtration plasmapheresis > immunoadsorption. One course consists of 5–6 sessions, and after treatment, hospitalization is required until the body’s IgG levels recover. Note that for “optic neuritis,” it may not be covered by insurance, and explanation to the patient is necessary.
In AQP4+ NMOSD, maintenance therapy should be initiated early after the first attack 1). In Japan, it is common to switch to prednisolone 5–10 mg/day plus azathioprine 50–100 mg/day after plasma exchange.
Biologics with high levels of evidence are as follows:
Complement inhibitors
Eculizumab: 900 mg IV weekly × 4 doses, then 1,200 mg every 2 weeks as maintenance 1)
Ravulizumab: Weight-based loading dose (2,400–3,000 mg) → 3,000–3,600 mg every 8 weeks from day 15 onward1)
B-cell depletion therapy
Rituximab: 375 mg/m² IV weekly × 4 doses, or 1,000 mg × 2 doses (2 weeks apart) → then 1,000 mg × 2 doses every 6 months1)
Inebilizumab: 300 mg IV twice every 15 days → every 6 months1)
IL-6 receptor inhibitor
Satralizumab: 120 mg subcutaneous injection every 4 weeks1)
QWhat if steroid pulse therapy is ineffective?
A
Plasma exchange therapy is the next option. Choose from simple plasma exchange, double filtration plasmapheresis, or immunoadsorption. Simple plasma exchange is considered most effective but also places a greater burden on the body. A course consists of 5–6 sessions, and hospitalization is required after treatment.
NMOSD is essentially an astrocytopathy. The pathogenesis is as follows.
Antibody production and blood-brain barrier (BBB) passage
In the periphery, B cells differentiate into AQP4-IgG-secreting plasmablasts. IL-6 promotes this differentiation and increases BBB permeability. The area postrema lacks a BBB and may serve as a route for AQP4-IgG entry into the CNS.
Cascade of astrocyte damage
AQP4-IgG binds to AQP4 water channels highly expressed on astrocyte foot processes, leading to astrocyte injury through the following pathways.
Classical complement pathway activation: The Fc portion of AQP4-IgG activates complement, forming the membrane attack complex (MAC) and directly damaging astrocytes.
ADCC (antibody-dependent cellular cytotoxicity): NK cells and neutrophils damage astrocytes via Fcγ receptors.
In MS, CD8+ T cells play a central role with predominant white matter demyelination, whereas in NMOSD, CD4+ T cells are more involved, forming necrotic lesions affecting both gray and white matter.
Reason for distribution
AQP4 channels are abundantly distributed in the optic nerve, area postrema, and spinal cord, making these regions selectively targeted.
Biomarkers
Serum GFAP: Reflects astrocyte damage and is elevated during attacks
Serum neurofilament light chain (NfL): Reflects axonal injury and correlates with attack severity1)
Cytokines involved include IL-6, IL-10, IL-17a, G-CSF, TNF-α, and BAFF/APRIL.
7. Latest Research and Future Prospects (Research Stage Reports)
It is estimated that 3–5% of NMOSD cases are paraneoplastic. Cases associated with ovarian teratoma have been studied in particular detail.
Ikeguchi et al. (2021) conducted a review of 6 cases of ovarian teratoma-associated AQP4+ NMOSD2). All cases were female, with a mean age of onset of 32.7 years (range 15–50 years). Among the 6 cases, 83% (5/6) presented with nausea and vomiting, 83% had positive CSF oligoclonal bands, and 83% had dorsal brainstem lesions. Pathological analysis revealed AQP4 immunoreactivity and lymphocytic infiltration in GFAP-positive neural tissue within the tumor, suggesting a mechanism where AQP4 antigen presentation within the tumor triggers an autoimmune response. After tumor resection, AQP4-IgG became negative in 60% (3/5) of cases.
Ding et al. (2021) conducted a review of 43 cases of paraneoplastic NMOSD3). 88.4% were female, and breast cancer and lung cancer were the most common tumor types. The importance of tumor screening is emphasized, especially in NMOSD patients aged 50 years or older.
Screening for tumors, including teratomas, is recommended even in younger patients.
The efficacy of Protein-A immunoadsorption (IA) therapy has been reported for refractory NMOSD that is unresponsive to steroids, plasma exchange, and rituximab.
Fan et al. (2024) performed three sessions of Protein-A immunoadsorption therapy in a 35-year-old woman with refractory NMOSD associated with Sjögren’s syndrome that did not respond to steroid pulse therapy and IVIG4). Within one week, visual impairment, paraplegia, and proprioceptive sensory deficits markedly improved, and rapid decreases in AQP4-IgG, IgA, IgG, and IgM were confirmed. No recurrence or progression was observed during 4 years of follow-up.
Characteristics of cases with comorbid autoimmune diseases
The actual status of various autoimmune diseases complicating NMOSD is becoming clearer.
Zhu et al. (2025) reported a case of a 14-year-old girl who developed NMOSD at age 115). She was AQP4-IgG positive, and primary Sjögren’s syndrome was confirmed during the course. Remission was maintained by switching from methylprednisolone, IVIG, and mycophenolate mofetil (MMF) to tacrolimus. Although autoimmune disease complications have been reported in 20–30% of adult NMOSD cases, this case shows that such complications also occur in pediatric patients5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, et al. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain Behav. 2021;11(9):e2282.
Fan W, Chen X, Xiao P, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Front Immunol. 2024;15:1429405.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.