Die Neuromyelitis-optica-Spektrum-Erkrankung (NMOSD) ist eine entzündliche, antikörpervermittelte Autoimmunerkrankung des zentralen Nervensystems. Früher auch als „Devic-Krankheit“ bekannt, wurde sie lange Zeit als Subtyp der Multiplen Sklerose (MS) angesehen. Mit der Entdeckung von Autoantikörpern gegen Aquaporin-4 (AQP4-IgG) im Jahr 2004 etablierte sie sich jedoch als eigenständige Krankheitseinheit.
Historischer Hintergrund: Im Jahr 1870 beschrieb Sir Thomas Clifford Allbutt erstmals den Zusammenhang zwischen Myelitis und Sehnervenstörungen. Spätere Forschungen erkannten sie als eine von der MS verschiedene Erkrankung, die heute unter dem Oberbegriff „NMOSD“ zusammengefasst wird.
Epidemiologie:
Inzidenz: Die geschätzte jährliche Inzidenz der AQP4+ NMOSD beträgt 0,4–7,3 pro 1.000.000 Personen1)
Geschlechterverteilung: Das Verhältnis von Männern zu Frauen beträgt etwa 1:9, mit einer deutlichen Betonung des weiblichen Geschlechts
Erkrankungsalter: Hauptsächlich im mittleren Alter (40–60 Jahre). In Japan liegt der Gipfel zwischen Ende 30 und Anfang 40
Ethnie: Häufiger bei Menschen afrikanischer und asiatischer Abstammung1)
Zusammenhang mit Schwangerschaft: Bei etwa 20–47 % der Frauen tritt die erste Episode während der Schwangerschaft oder innerhalb eines Jahres nach der Geburt oder Fehlgeburt auf.
QWie unterscheidet sich NMOSD von Multipler Sklerose (MS)?
A
NMOSD ist eine Antikörper-vermittelte Erkrankung, die auf AQP4-Wasserkanäle in Astrozyten abzielt. Pathologie, Behandlung und Prognose unterscheiden sich grundlegend von MS. Typisch für NMOSD sind LETM und schwere Optikusneuritis. Ein wichtiger Unterschied ist auch, dass krankheitsmodifizierende Medikamente wie Interferon-beta, die bei MS wirksam sind, bei NMOSD Rückfälle auslösen können.
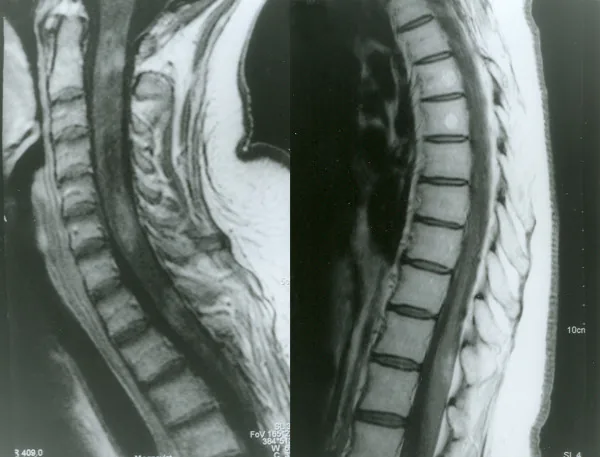
Kontrastmittel-MRT des Rückenmarks bei Neuromyelitis optica
Petelin Gadze Z, et al. Patient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case report. BMC Neurol. 2009. Figure 2. PMCID: PMC2773232. License: CC BY.
Gadolinium-verstärkte T1-gewichtete MRT-Aufnahme des Rückenmarks vom April 2007, die inhomogene ringförmige Kontrastmittelanreicherungen auf den Ebenen C2, C4-C5 und Th1 zeigt. Dies entspricht der Myelitis, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt wird.
Die Symptome der NMOSD variieren je nach betroffenem Bereich.
Akute Sehverschlechterung: Eines der Hauptsymptome. Charakteristisch ist die Resistenz gegen Steroidtherapie.
Augenschmerzen: Treten bei etwa der Hälfte der Fälle im Zusammenhang mit einer Optikusneuritis auf.
Farbsehstörungen: Typisch ist eine verminderte Rotsättigung.
Gesichtsfeldausfälle: Nicht nur zentrale Skotome, sondern auch horizontale Hemianopsie, bitemporale Hemianopsie und homonyme Hemianopsie können auftreten, da die Läsionen bis zum Chiasma opticum und Tractus opticus reichen.
Sensibilitätsstörungen und Paraparese: Motorische und sensorische Störungen durch Myelitis.
Blasen- und Mastdarmstörungen: Autonome Störungen im Zusammenhang mit Myelitis.
Therapieresistenter Schluckauf und Übelkeit/Erbrechen: Charakteristische Symptome durch Läsionen der Area postrema.
Augenbewegungsstörungen: Verursacht durch Hirnstammläsionen.
Hypersomnie (narkolepsieähnlich): aufgrund einer Läsion im Zwischenhirn/Hypothalamus
Zu Beginn der Erkrankung können grippeähnliche Symptome (Fieber, Muskelschmerzen, Kopfschmerzen) auftreten.
Beidseitig gleichzeitig: Die Optikusneuritis bei NMOSD ist in 17–82 % der Fälle beidseitig gleichzeitig. Wichtiger Unterschied zur MS.
Schwere Sehverschlechterung: Bei AQP4+ NMOSD liegt der mediane minimale Visus auf dem Niveau von Handbewegungen (HM). Auch nach Erholung beträgt der mediane Visus Fingerzählen, und 60–69 % behalten eine dauerhafte Sehbehinderung von mindestens 20/200 auf einem Auge.
Myelitis
LETM (longitudinally extensive transverse myelitis): Kontinuierliche Läsion über drei oder mehr Wirbelkörper. Etwa 85 % der AQP4+ NMOSD zeigen dies bei akuter Myelitis.
Komplettes Rückenmarksyndrom: Beteiligung aller drei Bahnen (motorisch, sensorisch, autonom).
Schwere funktionelle Beeinträchtigung: Über 30 % der Patienten sind im Nadir des Anfalls auf den Rollstuhl angewiesen. 37–44 % der AQP4+ NMOSD benötigen letztlich Gehhilfen.
Area-postrema-Syndrom
Therapieresistenter Schluckauf: Hält Tage bis Wochen an und spricht nicht auf übliche Antiemetika an.
Übelkeit und Erbrechen: Hintergrund ist, dass die Area postrema die Blut-Hirn-Schranke vermissen lässt, sodass AQP4-IgG dort leicht direkt angreifen kann.
Diagnostischer Kernbefund der NMOSD: Unerklärlicher therapieresistenter Schluckauf ist ein Anlass, aktiv an NMOSD zu denken.
QWie unterscheidet sich die Optikusneuritis bei NMOSD von der MS-assoziierten?
A
Die Optikusneuritis bei NMOSD ist schwerer, bilateral und rezidivierend, mit schlechterer Sehprognose. Bei AQP4+ NMOSD wird berichtet, dass 60–69 % der Patienten einen dauerhaften Sehverlust von 20/200 oder schlechter auf mindestens einem Auge erleiden. Zudem ist das Chiasma opticum häufiger betroffen, was zu verschiedenen Gesichtsfeldausfällen wie bitemporaler Hemianopsie führt – ein weiterer Unterschied zur MS.
Spinales MRT: LETM ist am charakteristischsten. Bevorzugt die zentrale graue Substanz. Begleitet von Rückenmarksschwellung, T1-Hypointensität und Gd-Anreicherung. Etwa 85 % der AQP4+ NMOSD-Patienten zeigen während einer akuten Myelitis eine LETM1)
Optikus-MRT: Fettsupprimierte Aufnahmen sind obligat. Charakteristisch sind bilaterale, langstreckige Entzündungen (>50 %). Beteiligung des hinteren Abschnitts und des Chiasmas ist typisch für AQP4+ NMOSD1)
Gehirn-MRT: Läsionen in der Area postrema, periventrikulären Hirnstammläsionen, hypothalamischen/periventrikulären Läsionen des dritten Ventrikels, ausgedehnte weiße Substanzläsionen usw.
Charakteristika der NMOSD: Im Gegensatz zur MS sind asymptomatische neue T2-Läsionen selten (3–13 %). Eine Überwachungs-MRT ist in der Regel nicht erforderlich1)
Bei älteren Patienten ist die Abgrenzung zur ischämischen Optikusneuropathie, zervikalen Myelopathie, spinalen Ischämie und primären ZNS-Lymphom wichtig.
QKann NMOSD auch bei negativem AQP4-Antikörper diagnostiziert werden?
A
Ja. Auch bei negativem oder nicht getestetem AQP4-IgG kann NMOSD diagnostiziert werden, wenn mindestens zwei der wichtigsten klinischen Merkmale erfüllt sind, zusätzliche MRT-Kriterien vorliegen und andere Erkrankungen ausgeschlossen wurden. Bei etwa 30 % der AQP4-IgG-negativen Fälle ist MOG-IgG positiv, daher wird die Bestimmung beider Antikörper empfohlen. Weniger als 1 % der AQP4-IgG-negativen Fälle konvertieren später zu positiv.
Methylprednisolon 1.000 mg/Tag intravenös über 3 Tage
Bei fehlender Sehverbesserung nach 3–4 Tagen Pause eine zweite Kur in Betracht ziehen.
Da die Optikusneuritis bei NMOSD resistent gegen Steroide ist, sollte bei unzureichendem Ansprechen frühzeitig die nächste Behandlung in Betracht gezogen werden.
Zweite Wahl: Plasmaaustauschtherapie
Wird durchgeführt, wenn keine Reaktion auf die Steroidpulstherapie erfolgt. Folgende Methoden stehen zur Auswahl.
Einfacher Plasmaaustausch (PE): Höchste Wirksamkeit, aber auch größte Belastung für den Körper
Doppelfiltrations-Plasmaaustausch (DFPP)
Immunadsorptionstherapie (IA): Ermöglicht selektive Entfernung von Antikörpern
Die Wirksamkeit wird in der Reihenfolge einfacher Plasmaaustausch > Doppelfiltration > Immunadsorption angegeben. Ein Zyklus umfasst 5–6 Sitzungen, und nach der Behandlung ist ein Krankenhausaufenthalt bis zur Erholung des IgG-Spiegels erforderlich. Beachten Sie, dass die Behandlung bei Optikusneuritis möglicherweise nicht von der Krankenkasse übernommen wird, was dem Patienten erklärt werden muss.
Bei AQP4+NMOSD sollte die Erhaltungstherapie früh nach dem ersten Anfall begonnen werden 1). In Japan ist nach Plasmaaustausch die Umstellung auf Prednisolon 5–10 mg/Tag plus Azathioprin 50–100 mg/Tag üblich.
Biologika mit hohem Evidenzgrad sind die folgenden:
Komplementinhibitoren
Eculizumab: 900 mg i.v. wöchentlich × 4, dann 1.200 mg alle 2 Wochen als Erhaltung 1)
Ravulizumab: gewichtsbasierte Aufsättigungsdosis (2.400–3.000 mg) → ab Tag 15 3.000–3.600 mg alle 8 Wochen 1)
B-Zell-Depletionstherapie
Rituximab: 375 mg/m² i.v. wöchentlich × 4 oder 1.000 mg × 2 (im Abstand von 2 Wochen) → alle 6 Monate 1.000 mg × 2 1)
Inebilizumab: 300 mg i.v. alle 15 Tage × 2 → alle 6 Monate 1)
IL-6-Rezeptor-Inhibitor
Satralizumab: 120 mg subkutane Injektion alle 4 Wochen1)
QWas tun, wenn die Steroid-Pulstherapie nicht wirkt?
A
Die Plasmapherese ist die nächste Option. Es stehen drei Verfahren zur Auswahl: einfacher Plasmaaustausch, doppelte Membranfiltration und Immunadsorption. Der einfache Plasmaaustausch gilt als am wirksamsten, belastet den Körper jedoch am stärksten. Ein Zyklus umfasst 5–6 Behandlungen, nach denen ein Krankenhausaufenthalt erforderlich ist.
6. Pathophysiologie und detaillierter Krankheitsmechanismus
NMOSD ist im Wesentlichen eine Astrozyten-Erkrankung (Astrozytopathie). Der Krankheitsmechanismus ist wie folgt:
Antikörperproduktion und Passage der Blut-Hirn-Schranke (BHS)
Peripher differenzieren B-Zellen zu AQP4-IgG-sezernierenden Plasmablasten. IL-6 fördert diese Differenzierung und erhöht die Permeabilität der BHS. Die Area postrema ist eine Region ohne BHS und kann als Eintrittspforte für AQP4-IgG in das ZNS dienen.
Kaskade der Astrozyten-Schädigung
AQP4-IgG bindet an AQP4-Wasserkanäle, die auf den Astrozytenfußfortsätzen hoch exprimiert werden, und führt über folgende Wege zur Astrozyten-Schädigung:
Aktivierung des klassischen Komplementwegs: Der Fc-Teil von AQP4-IgG aktiviert Komplement und bildet den Membranangriffskomplex (MAC), der Astrozyten direkt schädigt
ADCC (Antikörperabhängige zellvermittelte Zytotoxizität): NK-Zellen und Neutrophile schädigen Astrozyten über Fcγ-Rezeptoren
C5a-Anaphylatoxin-Freisetzung: Rekrutierung von Granulozyten (Neutrophile, Eosinophile), die sekundäre axonale Schädigung und Demyelinisierung verursachen 1)
Bei der MS steht die CD8+ T-Zell-vermittelte Demyelinisierung der weißen Substanz im Vordergrund, während bei der NMOSD CD4+ T-Zellen eine größere Rolle spielen und nekrotische Läsionen sowohl in der grauen als auch in der weißen Substanz bilden.
Grund der Verteilung
AQP4-Kanäle sind im Sehnerv, in der Area postrema und im Rückenmark stark exprimiert, sodass diese Regionen selektiv betroffen sind.
Biomarker
Serum-GFAP: Spiegelt die Astrozytenschädigung wider und ist während eines Schubs erhöht
Serum-Neurofilament-Leichtketten (NfL): Spiegeln axonale Schädigung wider und korrelieren mit der Schwere des Schubs 1)
Als beteiligte Zytokine wurden IL-6, IL-10, IL-17a, G-CSF, TNF-α und BAFF/APRIL beschrieben.
7. Aktuelle Forschung und zukünftige Perspektiven (Berichte aus der Forschungsphase)
Schätzungsweise 3–5 % der NMOSD-Fälle sind paraneoplastisch. Besonders gut untersucht ist der Zusammenhang mit Ovarialteratomen.
Ikeguchi et al. (2021) führten eine Übersicht über 6 Fälle von ovariellem Teratom-assoziierter AQP4+NMOSD durch2). Alle Patienten waren weiblich, das durchschnittliche Erkrankungsalter betrug 32,7 Jahre (15–50 Jahre). Von den 6 Fällen zeigten 83% (5/6) Übelkeit und Erbrechen, 83% hatten positive oligoklonale Banden im Liquor, und 83% wiesen dorsale Hirnstammläsionen auf. Die pathologische Analyse bestätigte AQP4-Immunreaktivität und lymphozytäre Infiltration in GFAP-positivem Nervengewebe innerhalb des Tumors, was darauf hindeutet, dass die AQP4-Antigenpräsentation im Tumor eine Autoimmunreaktion auslöst. Nach Tumorentfernung wurde bei 60% (3/5) der Fälle eine Serokonversion von AQP4-IgG zu negativ beobachtet.
Ding et al. (2021) führten eine Übersicht über 43 Fälle von paraneoplastischer NMOSD durch 3). 88,4 % waren weiblich, Brustkrebs und Lungenkrebs waren die häufigsten Tumortypen. Besonders bei NMOSD-Patienten über 50 Jahren wird die Bedeutung des Tumorscreenings betont.
Auch bei jungen Patienten wird ein Screening auf Tumoren einschließlich Teratomen empfohlen.
Immunadsorptionstherapie bei therapierefraktären Fällen
Für therapierefraktäre NMOSD, die nicht auf Steroide, Plasmaaustausch oder Rituximab ansprechen, wurde die Wirksamkeit der Protein-A-Immunadsorptionstherapie (IA) berichtet.
Fan et al. (2024) führten bei einer 35-jährigen Frau mit NMOSD und Sjögren-Syndrom, die nicht auf Steroidpulse und IVIG ansprach, drei Sitzungen Protein-A-Immunadsorption durch 4). Innerhalb einer Woche besserten sich Sehstörungen, Paraparese und Propriozeptionsstörungen deutlich, und ein rascher Abfall von AQP4-IgG, IgA, IgG und IgM wurde festgestellt. In einer vierjährigen Nachbeobachtung traten keine Rezidive oder Progression auf.
Merkmale von Fällen mit Begleiterkrankungen durch Autoimmunerkrankungen
Die tatsächliche Häufigkeit von Begleiterkrankungen durch verschiedene Autoimmunerkrankungen bei NMOSD wird zunehmend klarer.
Zhu et al. (2025) berichteten über den Fall eines 14-jährigen Mädchens, das im Alter von 11 Jahren an NMOSD erkrankte 5). Es war AQP4-IgG-positiv, und im Verlauf wurde eine begleitende primäre Sjögren-Syndrom diagnostiziert. Durch die Umstellung von Methylprednisolon, IVIG und Mycophenolatmofetil (MMF) auf Tacrolimus konnte eine Remission aufrechterhalten werden. Bei 20–30 % der erwachsenen NMOSD-Fälle wurde eine Begleiterkrankung durch Autoimmunerkrankungen berichtet, aber es wurde gezeigt, dass solche Begleiterkrankungen auch bei Kindern auftreten können 5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, et al. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain Behav. 2021;11(9):e2282.
Fan W, Chen X, Xiao P, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Front Immunol. 2024;15:1429405.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.