Le trouble du spectre de la neuromyélite optique (NMOSD) est une maladie inflammatoire, auto-immune et médiée par des anticorps qui affecte le système nerveux central. Autrefois appelée « maladie de Devic », elle a longtemps été considérée comme un sous-type de la sclérose en plaques (SEP). Cependant, la découverte en 2004 d’auto-anticorps dirigés contre l’aquaporine-4 (AQP4-IgG) a permis de l’établir comme une entité pathologique distincte.
Contexte historique : En 1870, Sir Thomas Clifford Allbutt a décrit pour la première fois l’association entre myélite et troubles optiques. Des recherches ultérieures ont permis de la reconnaître comme une maladie différente de la SEP, et elle est aujourd’hui appréhendée sous le concept global de « NMOSD ».
Épidémiologie :
Incidence : L’incidence annuelle estimée de la NMOSD AQP4+ est de 0,4 à 7,3 par million d’habitants1)
Sexe : Le ratio hommes/femmes est d’environ 1:9, avec une nette prédominance féminine
Âge de début : Survient principalement chez les adultes d’âge moyen (40-60 ans). Au Japon, le pic se situe entre la fin de la trentaine et le début de la quarantaine
Ethnie : Tendance à être plus fréquente chez les personnes d’origine africaine et asiatique1)
Association avec la grossesse : environ 20 à 47 % des femmes présentent une première poussée pendant la grossesse ou dans l’année suivant l’accouchement ou une fausse couche.
QQuelle est la différence entre la NMOSD et la sclérose en plaques (SEP) ?
A
La NMOSD est une maladie auto-immune médiée par des anticorps ciblant le canal hydrique AQP4 des astrocytes, dont la physiopathologie, le traitement et le pronostic diffèrent fondamentalement de ceux de la SEP. La NMOSD se caractérise typiquement par une LETM ou une névrite optique sévère, et il est important de noter que les traitements de fond efficaces dans la SEP, comme l’interféron bêta, peuvent déclencher des poussées de NMOSD.
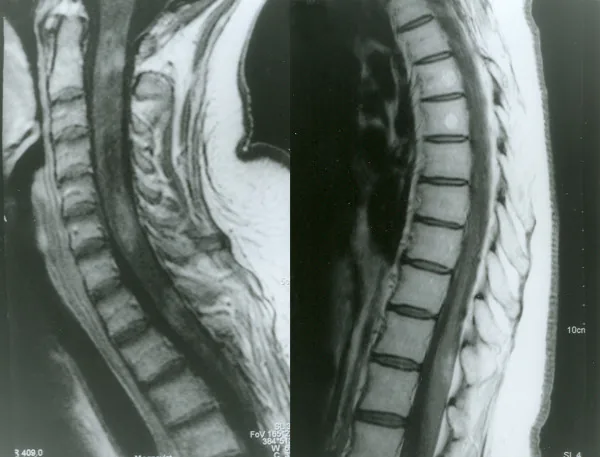
IRM médullaire avec contraste dans la neuromyélite optique
Petelin Gadze Z, et al. Patient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case report. BMC Neurol. 2009. Figure 2. PMCID: PMC2773232. License: CC BY.
Image IRM pondérée en T1 avec gadolinium de la moelle épinière réalisée en avril 2007, montrant un rehaussement annulaire hétérogène aux niveaux C2, C4-C5 et Th1, correspondant à la myélite traitée dans la section « 2. Principaux symptômes et signes cliniques ».
Les symptômes de la NMOSD varient selon la région atteinte.
Baisse brutale de l’acuité visuelle : l’un des principaux symptômes. Caractérisée par une résistance au traitement par corticoïdes.
Douleur oculaire : associée à la névrite optique, présente dans environ la moitié des cas.
Troubles de la vision des couleurs : diminution typique de la saturation du rouge.
Déficit du champ visuel : peut aller au-delà d’un scotome central, incluant une hémianopsie horizontale, bitemporale ou homonyme, en raison de l’atteinte du chiasma et du tractus optique.
Troubles sensitifs et paraplégie : troubles moteurs et sensitifs dus à la myélite.
Troubles vésico-sphinctériens : troubles autonomes associés à la myélite.
Hoquet incoercible et nausées/vomissements : symptômes caractéristiques d’une lésion de l’area postrema.
Troubles oculomoteurs : dus à une atteinte du tronc cérébral.
Hypersomnie (narcoleptique) : due à une lésion diencéphalique/hypothalamique
Au début de la maladie, des symptômes pseudo-grippaux (fièvre, myalgies, céphalées) peuvent apparaître.
Bilatéral simultané : la névrite optique dans la NMOSD est bilatérale simultanée dans 17 à 82 % des cas. Différence importante avec la SEP.
Baisse sévère de l’acuité visuelle : dans la NMOSD AQP4+, l’acuité visuelle minimale médiane est au niveau de la perception lumineuse (PL). Après récupération, l’acuité visuelle médiane reste au niveau de la perception des mouvements de la main (PM), et 60 à 69 % des patients conservent une déficience visuelle permanente d’au moins 20/200 dans un œil.
Myélite
LETM (myélite longitudinale étendue transverse) : lésion continue s’étendant sur au moins 3 vertèbres. Environ 85% des NMOSD AQP4+ la présentent lors d’une myélite aiguë.
Syndrome médullaire complet : implique les trois voies motrice, sensitive et autonome.
Handicap fonctionnel sévère : plus de 30% des patients sont dépendants du fauteuil roulant au nadir de la crise. 37 à 44% des NMOSD AQP4+ nécessitent finalement une aide à la marche.
Syndrome de l'area postrema
Hoquet réfractaire : persiste de plusieurs jours à plusieurs semaines, ne répond pas aux antiémétiques habituels.
Nausées et vomissements : l’area postrema manque de barrière hémato-encéphalique, ce qui permet aux AQP4-IgG d’y accéder directement.
Signe diagnostique clé du NMOSD : un hoquet réfractaire inexpliqué doit faire suspecter activement un NMOSD.
QEn quoi la névrite optique du NMOSD diffère-t-elle de celle liée à la SEP ?
A
La névrite optique du NMOSD est plus sévère, bilatérale et récurrente, avec un pronostic visuel défavorable. Dans le NMOSD AQP4+, 60 à 69 % des patients conservent une déficience visuelle permanente d’au moins 20/200 dans un œil. De plus, l’atteinte fréquente du chiasma optique entraîne divers troubles du champ visuel, comme une hémianopsie bitemporale, ce qui la distingue également de la SEP.
Les critères diagnostiques de la NMOSD avec AQP4-IgG positive sont les trois éléments suivants :
Au moins une caractéristique clinique majeure
AQP4-IgG positive (en utilisant la meilleure méthode de détection)
Exclusion d’autres diagnostics
Les critères diagnostiques du NMOSD pour les patients AQP4-IgG négatifs ou non testés sont les suivants : les 4 éléments ci-dessous doivent être remplis.
Au moins 2 caractéristiques cliniques majeures (dont l’une doit être une névrite optique, une LETM ou un syndrome de l’area postrema)
Dissémination spatiale
Respect des critères IRM supplémentaires
Exclusion d’autres diagnostics
Les caractéristiques cliniques majeures (6 éléments) sont les suivantes :
Névrite optique
Myélite aiguë
Syndrome de la dernière aire (hoquet réfractaire, nausées et vomissements)
Syndrome aigu du tronc cérébral
Narcolepsie symptomatique / syndrome aigu de l’hypothalamus
IRM médullaire : la LETM est la plus caractéristique. Prédominance de la substance grise centrale. Accompagnée d’un gonflement médullaire, d’un hyposignal T1 et d’une prise de contraste Gd. Environ 85 % des NMOSD AQP4+ présentent une LETM lors de la myélite aiguë1)
IRM du nerf optique : les séquences avec saturation de graisse sont essentielles. Inflammation bilatérale et étendue (>50 %) caractéristique. L’atteinte de la partie postérieure et du chiasma est typique de la NMOSD AQP4+1)
IRM cérébrale : lésions de l’area postrema, lésions du tronc cérébral autour du 4e ventricule, lésions hypothalamiques/périventriculaires du 3e ventricule, lésions étendues de la substance blanche, etc.
Caractéristiques de la NMOSD : contrairement à la SEP, les nouvelles lésions T2 asymptomatiques sont rares (3 à 13 %). L’IRM de surveillance n’est généralement pas nécessaire1)
Chez les personnes âgées, il est important de différencier la neuropathie optique ischémique, la myélopathie cervicarthrosique, l’infarctus médullaire et le lymphome primitif du système nerveux central.
QPeut-on diagnostiquer une NMOSD même si les anticorps AQP4 sont négatifs ?
A
Oui. Même en l’absence d’anticorps AQP4-IgG ou sans test, le diagnostic de NMOSD peut être posé si au moins deux caractéristiques cliniques principales sont présentes, les critères IRM supplémentaires sont remplis et les autres maladies sont exclues. De plus, environ 30 % des cas négatifs pour les anticorps AQP4-IgG sont positifs pour les anticorps MOG-IgG, et le dosage des deux anticorps est recommandé. Moins de 1 % des cas négatifs pour les anticorps AQP4-IgG présentent une séroconversion ultérieure.
Première intention : traitement par bolus de stéroïdes
Administrer 1 000 mg/jour de méthylprednisolone par perfusion intraveineuse pendant 3 jours.
Si l’acuité visuelle ne s’améliore pas, envisager une nouvelle cure après un intervalle de 3 à 4 jours.
La névrite optique dans la NMOSD est très résistante aux stéroïdes, donc si la réponse est insuffisante, envisager rapidement le traitement suivant.
Deuxième intention : plasmaphérèse
Réalisée en cas d’absence de réponse aux bolus de stéroïdes. Les options suivantes sont disponibles.
Plasmaphérèse simple (PE) : la plus efficace mais aussi la plus agressive pour l’organisme.
Plasmaphérèse par double filtration (DFPP)
Immunoadsorption (IA) : permet l’élimination sélective des anticorps.
L’ordre d’efficacité est : plasmaphérèse simple > double filtration > immunoadsorption. Un cycle de 5 à 6 séances est réalisé, et une hospitalisation est nécessaire jusqu’à la récupération du taux d’IgG sérique. À noter que pour la « névrite optique », ce traitement peut ne pas être couvert par l’assurance maladie, ce qui nécessite une explication au patient.
Dans la NMOSD AQP4+, un traitement d’entretien doit être débuté précocement après la première poussée 1). Au Japon, après échange plasmatique, le passage à la prednisolone 5–10 mg/jour + azathioprine 50–100 mg/jour est courant.
Les agents biologiques avec un haut niveau de preuve sont les suivants :
Inhibiteurs du complément
Éculizumab : 900 mg IV par semaine × 4, puis 1 200 mg toutes les 2 semaines en entretien 1)
Ravulizumab : dose de charge basée sur le poids (2 400–3 000 mg), puis 3 000–3 600 mg toutes les 8 semaines à partir du jour 15 1)
Thérapie de déplétion des cellules B
Rituximab : 375 mg/m² IV par semaine × 4, ou 1 000 mg × 2 (à 2 semaines d’intervalle), puis 1 000 mg × 2 tous les 6 mois 1)
Inebilizumab : 300 mg IV toutes les 2 semaines × 2, puis tous les 6 mois 1)
Inhibiteur du récepteur de l’IL-6
Satralizumab : 120 mg en injection sous-cutanée toutes les 4 semaines1)
QQue faire si la corticothérapie par bolus est inefficace ?
A
L’échange plasmatique est l’option suivante. Il existe trois types : échange plasmatique simple, plasmaphérèse à double filtration et immunoadsorption. L’échange plasmatique simple est considéré comme le plus efficace mais aussi le plus lourd pour l’organisme. Une cure comprend 5 à 6 séances, et une hospitalisation est nécessaire après le traitement.
La NMOSD est essentiellement une astrocytopathie. Les mécanismes sont les suivants :
Production d’anticorps et passage de la barrière hémato-encéphalique (BHE)
En périphérie, les cellules B se différencient en plasmablastes sécrétant l’AQP4-IgG. L’IL-6 favorise cette différenciation et augmente la perméabilité de la BHE. L’area postrema est une région dépourvue de BHE et peut servir de voie d’entrée de l’AQP4-IgG dans le SNC.
Chaîne de lésions astrocytaires
L’AQP4-IgG se lie aux canaux hydriques AQP4 fortement exprimés sur les pieds astrocytaires, entraînant des lésions astrocytaires par les voies suivantes :
Activation de la voie classique du complément : La partie Fc de l’AQP4-IgG active le complément, formant le complexe d’attaque membranaire (MAC) qui lèse directement les astrocytes
ADCC (cytotoxicité à médiation cellulaire dépendante des anticorps) : Les cellules NK et les neutrophiles lèsent les astrocytes via les récepteurs Fcγ
Libération de l’anaphylatoxine C5a : Recrute les granulocytes (neutrophiles et éosinophiles), provoquant des lésions axonales secondaires et une démyélinisation 1)
Dans la SEP, la démyélinisation est principalement centrée sur la substance blanche avec un rôle central des lymphocytes T CD8+, tandis que dans la NMOSD, les lymphocytes T CD4+ jouent un rôle majeur, formant des lésions nécrotiques touchant à la fois la substance grise et la substance blanche.
Raison de la distribution
Les canaux AQP4 sont abondamment distribués dans le nerf optique, l’area postrema et la moelle épinière, ce qui explique pourquoi ces régions sont sélectivement ciblées.
Biomarqueurs
GFAP sérique : Reflète les lésions astrocytaires, élevée lors des poussées
Chaîne légère de neurofilament sérique (NfL) : Reflète les lésions axonales, corrélée à la sévérité des poussées 1)
Les cytokines impliquées rapportées sont IL-6, IL-10, IL-17a, G-CSF, TNF-α et BAFF/APRIL.
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
On estime que 3 à 5 % des NMOSD sont paranéoplasiques. Les cas associés au tératome ovarien ont été particulièrement étudiés en détail.
Ikeguchi et al. (2021) ont réalisé une revue de 6 cas de NMOSD AQP4+ associée au tératome ovarien2). Tous les cas étaient des femmes, avec un âge moyen d’apparition de 32,7 ans (15 à 50 ans). Parmi les 6 cas, 83 % (5/6) présentaient des nausées et vomissements, 83 % avaient des bandes oligoclonales positives dans le LCR, et 83 % présentaient des lésions du tronc cérébral dorsal. L’analyse pathologique a confirmé une immunoréactivité AQP4 et une infiltration lymphocytaire dans le tissu nerveux GFAP-positif de la tumeur, suggérant un mécanisme par lequel la présentation de l’antigène AQP4 dans la tumeur déclenche une réaction auto-immune. Après résection tumorale, l’AQP4-IgG s’est négativée chez 60 % (3/5) des cas.
Ding et al. (2021) ont réalisé une revue de 43 cas de NMOSD paranéoplasique3). 88,4 % étaient des femmes, et les cancers du sein et du poumon étaient les types tumoraux les plus fréquents. L’importance du dépistage tumoral est soulignée, en particulier chez les patients atteints de NMOSD âgés de plus de 50 ans.
Même chez les jeunes patients, un dépistage tumoral incluant les tératomes est recommandé.
Traitement par immunoadsorption pour les cas réfractaires
L’efficacité de l’immunoadsorption sur protéine A (IA) a été rapportée dans les NMOSD réfractaires ne répondant pas aux stéroïdes, à la plasmaphérèse ou au rituximab.
Fan et al. (2024) ont réalisé 3 séances d’immunoadsorption sur protéine A chez une femme de 35 ans atteinte de NMOSD associée au syndrome de Sjögren, réfractaire aux bolus de stéroïdes et aux IVIG4). En une semaine, les troubles visuels, la paraplégie et les troubles proprioceptifs se sont nettement améliorés, avec une diminution rapide des AQP4-IgG, IgA, IgG et IgM. Aucune récidive ni progression n’a été observée pendant 4 ans de suivi.
Caractéristiques des cas associés à des maladies auto-immunes
La réalité des associations entre NMOSD et diverses maladies auto-immunes est de mieux en mieux comprise.
Zhu et al. (2025) ont rapporté le cas d’une fille de 14 ans ayant développé une NMOSD à l’âge de 11 ans5). AQP4-IgG positive, l’évolution a confirmé l’association à un syndrome de Sjögren primaire. La rémission a été maintenue par méthylprednisolone, IVIG, mycophénolate mofétil (MMF) puis passage au tacrolimus. Alors que 20 à 30 % des cas adultes de NMOSD présentent une association à une maladie auto-immune, cette observation montre que cette association existe aussi chez l’enfant5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, et al. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain Behav. 2021;11(9):e2282.
Fan W, Chen X, Xiao P, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Front Immunol. 2024;15:1429405.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.