La maladie de Behçet est une maladie inflammatoire systémique réfractaire d’origine inconnue, caractérisée par quatre symptômes principaux : ulcères aphteux buccaux, lésions oculaires, ulcères génitaux et lésions cutanées. L’inflammation est aiguë et transitoire, mais la maladie se caractérise par des récidives répétées. Elle survient chez les adultes jeunes et d’âge moyen (20-50 ans), avec un ratio hommes-femmes d’environ 1:1. L’uvéite survient chez environ 70 % des hommes et 45 % des femmes, et les cas graves sont plus fréquents chez les jeunes hommes.
La maladie est fréquente le long de la route de la soie, de la Méditerranée à la Chine et au Japon. Environ 50 % des patients sont positifs pour HLA-B51 (contre 15 % dans la population générale)5), et HLA-A26 (A*2601) a également été rapporté comme un allèle de risque indépendant. On pense que des prédispositions immunogénétiques et des facteurs environnementaux sont impliqués dans le déclenchement de la maladie. Autrefois, cette maladie avait un taux élevé de cécité, mais grâce aux progrès des immunosuppresseurs et des agents biologiques, le taux de cécité a diminué.
La maladie de Behçet est enregistrée comme maladie rare désignée n°568).
Caractéristiques de la maladie
Âge d’apparition : Adultes jeunes et d’âge moyen (20-50 ans)
Sexe : Ratio hommes-femmes environ 1:1. Les lésions oculaires sont plus graves chez les jeunes hommes.
Distribution géographique : Fréquente le long de la route de la soie (Méditerranée, Moyen-Orient, Asie de l’Est)
Contexte génétique : Positivité HLA-B51 (environ 50 % des patients), HLA-A26 également un risque indépendant5)
4 symptômes principaux
Ulcères aphteux buccaux : ulcères récurrents douloureux. Souvent le premier symptôme.
La proportion de la maladie de Behçet parmi l’ensemble des uvéites tend à diminuer avec le temps. Selon une enquête épidémiologique de 2002, elle était de 6,2 % (3e rang), mais en 2009, elle est tombée à 3,9 % (6e rang)5). Outre la diminution du nombre de patients, un allègement de la maladie a également été rapporté4).
QQu'est-ce que la maladie de Behçet ?
A
La maladie de Behçet est une maladie inflammatoire systémique récurrente caractérisée par quatre symptômes principaux : ulcères aphteux buccaux, symptômes oculaires, ulcères génitaux et symptômes cutanés. La cause est inconnue, mais un mécanisme auto-immun est suspecté. Elle est fréquente le long de la Route de la Soie et impliquerait des facteurs génétiques (HLA-B51) et environnementaux. Les lésions oculaires sont particulièrement importantes et peuvent conduire à la cécité en l’absence de traitement approprié.
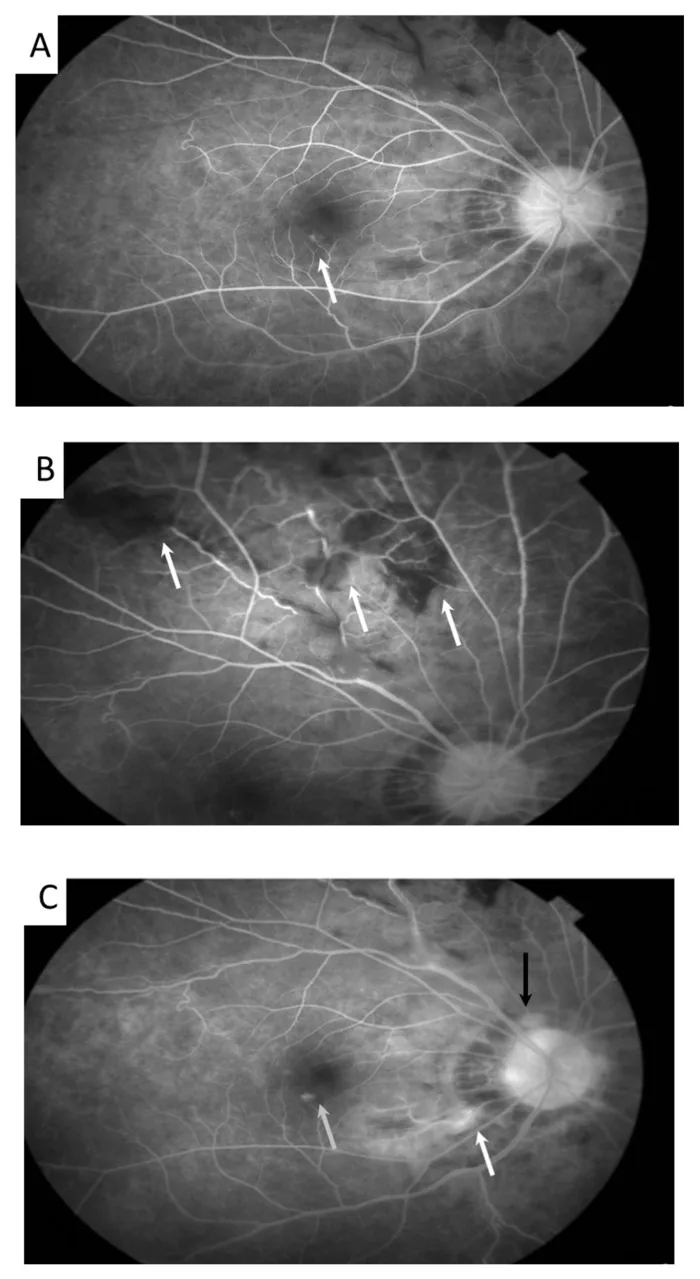
Meng PP, et al. Use of Ultra-Widefield Fluorescein Angiography to Guide the Treatment to Idiopathic Retinal Vasculitis, Aneurysms, and Neuroretinitis-Case Report and Literature Review. Medicina (Kaunas). 2022. Figure 2. PMCID: PMC9611749. License: CC BY.
Sur l’image en autofluorescence, on observe des dilatations anévrismales des artérioles rétiniennes (flèches) et une tortuosité veineuse. Cela correspond aux signes de vascularite rétinienne traités dans la section « 2. Principaux symptômes et signes cliniques ».
Les symptômes subjectifs oculaires comprennent un brouillard visuel, une baisse de l’acuité visuelle et des myodésopsies pendant les poussées inflammatoires. L’inflammation récidive souvent à des moments différents dans chaque œil, et environ 90 % des cas deviennent bilatéraux. Dans les cas graves, la fréquence des crises peut atteindre plusieurs fois par mois, tandis que dans les cas légers, une crise par an environ peut persister pendant plusieurs années, voire plus de dix ans. Des rétinites localisées à la macula peuvent se répéter, entraînant une baisse irréversible de l’acuité visuelle.
Les symptômes systémiques comprennent principalement des ulcères aphteux buccaux (environ 90 %), des symptômes cutanés (environ 75 %) et des ulcères génitaux (environ 50 %). Les ulcères aphteux buccaux surviennent fréquemment sur la langue, la muqueuse jugale, les lèvres et les gencives ; ce sont des ulcères douloureux entourés d’un érythème. Ils sont presque constants dans cette maladie et constituent souvent le premier symptôme. Ils guérissent sans cicatrice en moins de 10 jours, mais récidivent.
Comme symptômes secondaires, on observe une arthrite sans déformation ni raideur, une épididymite, une maladie de Behçet gastro-intestinale (ulcère iléo-cæcal), une maladie de Behçet vasculaire (vascularite) et une maladie de Behçet neurologique (encéphalomyélite). Les symptômes extra-oculaires et les signes oculaires ne sont pas toujours corrélés, il est donc important d’évaluer l’état général en plus de l’observation ophtalmologique3).
Signes précoces et de phase active de la crise oculaire : Inflammation oculaire paroxystique. Fins précipités rétro-cornéens, cellules inflammatoires dans la chambre antérieure et hypopyon. L’exsudation de fibrine dans la chambre antérieure est généralement absente. Pas de nodules iriens ou goniosynéchies (non granulomateux). Au segment postérieur, l’opacité vitréenne, les plaques exsudatives rétiniennes et les hémorragies disparaissent relativement rapidement (1 à 2 semaines), ce qui est caractéristique3).
Signes tardifs3) : Atrophie rétino-choroïdienne, blanchiment des vaisseaux rétiniens, pâleur de la papille optique → conduisant à une baisse sévère de la fonction visuelle.
L’hypopyon de la maladie de Behçet est principalement dû à une infiltration de neutrophiles, il est donc fluide et forme un niveau horizontal net. Il se déplace facilement avec les changements de position, ce qui le distingue de l’hypopyon visqueux de l’uvéite associée au HLA-B273). Les précipités rétro-cornéens sont fins et non granulomateux.
Lors des exacerbations aiguës de l’uvéite postérieure, on observe une opacité vitréenne, une rétinovasculite, des plaques exsudatives rétiniennes blanches et des hémorragies. Les plaques exsudatives blanches sont dues à une infiltration leucocytaire et à un gonflement des fibres nerveuses optiques lié à l’ischémie, et disparaissent relativement rapidement (en environ une semaine). La vascularite occlusive peut également provoquer des hémorragies ressemblant à une occlusion de branche veineuse rétinienne.
L’angiographie à la fluorescéine montre souvent, même en l’absence de crise oculaire, une fuite intense et étendue de fluorescéine au niveau des capillaires rétiniens (fuite en forme de fougère), considérée comme caractéristique de cette maladie.
BOS24 (Score de crise oculaire de la maladie de Behçet 24) 6)
Score quantifiant le degré d’inflammation pour chaque crise oculaire. Il comprend 6 items : inflammation de la chambre antérieure (max 4 points), opacité du vitré (max 4 points), lésions rétiniennes périphériques (max 8 points), lésions rétiniennes du pôle postérieur (max 4 points), lésions fovéolaires (max 2 points) et lésions du nerf optique (max 2 points), pour un total de 24 points. Utilisé pour l’évaluation objective de la sévérité des crises et pour juger de l’efficacité thérapeutique 6).
De plus, l’inflammation intraoculaire peut entraîner une cataracte compliquée et un glaucome secondaire, pouvant causer une baisse de l’acuité visuelle.
Signes cutanés : érythème noueux prédominant aux membres inférieurs, thrombophlébite sous-cutanée, éruptions folliculaires ou acnéiformes sur le visage, le cou et le dos. Les ulcères génitaux sont des ulcères aphteux douloureux aux bords nets, touchant le scrotum et le pénis chez l’homme, et les grandes et petites lèvres chez la femme.
Chez les enfants, des symptômes oculaires sévères incluant une uvéite postérieure, une vascularite rétinienne et une papillite ont été rapportés 1), avec un retard diagnostique pouvant atteindre en moyenne 11,3 ± 8,5 mois 1).
QComment distinguer l'hypopyon de la maladie de Behçet ?
A
L’hypopyon de la maladie de Behçet est de nature neutrophilique, donc « fluide », formant un niveau horizontal net et se déplaçant avec les changements de position. En revanche, l’hypopyon de l’uvéite associée au HLA-B27 est plus visqueux, ne forme pas de niveau et a un aspect surélevé 3). Cette différence de consistance est un indice diagnostique.
La cause de la maladie de Behçet est inconnue, mais on suppose que des micro-organismes pathogènes tels que les streptocoques sont impliqués comme facteurs externes, et qu’une prédisposition génétique et des anomalies immunitaires sont impliquées comme facteurs internes. Les anomalies fonctionnelles des neutrophiles et les anomalies des cytokines, notamment le TNF-α, jouent un rôle central, entraînant des réactions inflammatoires paroxystiques et récurrentes principalement au niveau de la muqueuse buccale, des yeux, de la peau et des organes génitaux externes.
Facteurs de risque
Description
HLA-B51
Positif chez environ 50 % des patients (15 % dans la population générale). Principal marqueur génétique 5)
HLA-A26
Signalé comme allèle de risque indépendant 2)
Région / ethnie
Fréquent le long de la Route de la Soie (Méditerranée, Moyen-Orient, Asie de l’Est)
Sexe / âge
Apparition entre 20 et 50 ans. Lésions oculaires sévères fréquentes chez les jeunes hommes
Chez les enfants, cela représente environ 1,6 à 7,7 % de tous les cas de maladie de Behçet2), et une aggravation des lésions oculaires a été rapportée chez les cas HLA-B51 positifs1).
:::tip Conseils pour la vie quotidienne
La colchicine doit être prise à long terme pour prévenir les crises oculaires. Même si les crises oculaires semblent s’être calmées, n’arrêtez pas le traitement de votre propre chef. De plus, comme on sait que le traitement par photocoagulation peut déclencher des crises sévères, il est important d’en discuter suffisamment avec votre médecin traitant.
:::
Le diagnostic de la maladie de Behçet est basé sur les directives cliniques du groupe d’étude de la maladie de Behçet du ministère de la Santé, du Travail et des Affaires sociales (1987, révision mineure en 2016). Pour le diagnostic basé sur les symptômes oculaires, se référer aux directives cliniques pour les lésions oculaires de la maladie de Behçet (2012) 3).
Le type complet est défini par l’apparition des quatre symptômes principaux au cours de l’évolution, et le type incomplet par l’apparition de trois symptômes principaux ou de deux symptômes principaux et deux symptômes secondaires (ou des symptômes oculaires typiques avec un autre symptôme principal et deux symptômes secondaires).
Cinq symptômes secondaires (révision mineure de 2016)8) : (1) arthrite sans déformation ni raideur, (2) épididymite, (3) lésions gastro-intestinales représentées par l’ulcère iléo-cæcal, (4) lésions vasculaires, (5) lésions du système nerveux central modérées à sévères.
Type
Critères
Type complet
Apparition des quatre symptômes principaux
Type incomplet
Trois symptômes principaux ou deux symptômes principaux + deux symptômes secondaires
Type spécial
Type intestinal, vasculaire ou neurologique (lorsque les critères de forme complète ou incomplète sont remplis et qu’il existe des lésions spéciales)
Les critères diagnostiques internationaux du Groupe d’étude international sur la maladie de Behçet (critères ISG, 1990) 7) sont également largement utilisés dans le monde.
Analyses sanguines : numération leucocytaire, CRP, VS (marqueurs inflammatoires)
Test de pathergie cutanée : érythème et formation de pustules après piqûre intradermique (diagnostic auxiliaire)
Chez les enfants, le diagnostic est souvent retardé ; une étude rapporte un délai moyen de 11,3 ± 8,5 mois entre l’apparition des symptômes oculaires et le diagnostic1).
Uvéite associée au HLA-B27 : hypopyon visqueux, de forme irrégulière avec un centre légèrement surélevé. L’inflammation du segment postérieur est presque absente3). Endophtalmie fongique : forme un hypopyon et une opacité du vitré, mais progressive. Iritis diabétique : forme rarement un niveau liquide3).
Colchicine orale (hors AMM) : 0,5 à 1,5 mg/jour, généralement 1 mg/jour en deux prises. Une amélioration partielle est observée chez environ 60 % des patients3). Effets secondaires : symptômes gastro-intestinaux tels que diarrhée, tératogénicité. Même après la stabilisation des crises oculaires, un traitement à long terme est nécessaire, avec des analyses sanguines régulières (surveillance des atteintes hépatiques et rénales, granulopénie, rhabdomyolyse).
Étape 3 : En cas de réponse insuffisante à la colchicine
Ciclosporine (Neoral® 50 mg) : 5 mg/kg/jour en deux prises. Surveillance avec un objectif de concentration résiduelle de 50 à 200 ng/mL.
:::caution Effets secondaires importants de la ciclosporine
Une insuffisance rénale survient fréquemment. De plus, l’apparition d’une maladie de Neuro-Behçet est observée chez environ 20 % des patients, et une attention particulière aux symptômes neurologiques est nécessaire en cas d’administration à long terme3). L’association avec la colchicine comporte un risque de myopathie.
:::
Étape 4 : Inhibiteurs du TNF (cas réfractaires et sévères)
Approbation : Approuvé en 2007 pour l’uvéite rétinienne réfractaire associée à la maladie de Behçet
Indication : uvéite rétinienne réfractaire résistante au traitement existant, ou lorsque l’utilisation d’immunosuppresseurs est difficile en raison d’effets secondaires systémiques
Posologie : 5 mg/kg en perfusion intraveineuse sur au moins 2 heures. Administration à 2 et 6 semaines après la première dose, puis toutes les 8 semaines. Administrer à travers un filtre membrane de 1,2 μm ou moins. Après la 6e semaine, si aucune réaction à la perfusion n’est observée, la durée de la perfusion peut être réduite (à condition que le débit moyen ne dépasse pas 5 mg/kg par heure)
Échec primaire et secondaire : il existe un échec primaire (aucun effet dès le début) et un échec secondaire (diminution de l’efficacité en cours de traitement)
Dépistage pré-administration (directives d’utilisation des inhibiteurs du TNF)9) :
Radiographie thoracique, test cutané à la tuberculine, et en cas de forte positivité, QFT/T-SPOT
Lésions tuberculeuses anciennes → prise d’isoniazide de 3 semaines avant le début de l’infliximab jusqu’au 9e mois
HBsAg, anticorps anti-HBs, anticorps anti-HBc (surveillance de la réactivation du VHB)
HBsAg positif → consultation obligatoire avec un spécialiste hépatologue. Même négatif, si anticorps anti-HBs/anti-HBc positifs (infection antérieure) → mesure régulière de l’ADN du VHB
Vérification de la présence d’hépatite C, HTLV-1 et infection par le VIH
Contre-indications9) : infections incluant la tuberculose active (infections mycobactériennes atypiques, infection par le virus de l’hépatite B), insuffisance cardiaque congestive (NYHA classe III ou plus), tumeur maligne, maladie démyélinisante (sclérose en plaques, etc.)
Surveillance des effets secondaires9) : analyses sanguines périphériques régulières (globules blancs, lymphocytes) et biochimiques (incluant CRP). Attention à l’apparition de tuberculose et de pneumocystose (radiographie thoracique, scanner, β-D-glucane). Attention à la réactivation d’une infection antérieure par le virus de l’hépatite B (ADN-VHB). Réactions à la perfusion (observation pendant la perfusion et 2 heures après). Attention également aux réactions d’hypersensibilité retardée (douleurs musculaires, éruption cutanée, fièvre, douleurs articulaires survenant plus de 3 jours après la perfusion).
Critères pour le médecin et l’établissement9) : spécialiste certifié par la Société japonaise d’ophtalmologie et membre de la Société japonaise d’uvéite, avoir terminé l’apprentissage en ligne de cette société. L’établissement d’introduction doit être enregistré auprès de la Société japonaise d’uvéite. Une capacité à gérer les effets secondaires graves, à prendre en charge les problèmes respiratoires/infectieux, et une collaboration avec un interniste familier avec les anti-TNF sont requises.
Cas représentatif : homme de 32 ans, forme complète HLA-B51 positif. Difficulté de contrôle avec cyclosporine, prednisolone et colchicine → introduction d’infliximab. 3 poussées oculaires au total sur les deux yeux dans l’année précédant le début → disparition des poussées dans l’année suivant le début. Acuité visuelle corrigée : droite 1,2/gauche 0,7 → amélioration à droite 1,2/gauche 0,9. Maintien d’une bonne acuité visuelle après 3 ans et 6 mois3).
Approbation de l’assurance maladie : approuvé en 2016 pour les uvéites non infectieuses intermédiaires, postérieures et panuvéites
Posologie : 80 mg en injection sous-cutanée initialement → 40 mg une semaine plus tard → puis 40 mg toutes les deux semaines en injection sous-cutanée
Auto-injection : possible après une formation et un entraînement suffisants. Une surveillance des signes oculaires et systémiques tous les 2 à 3 mois est obligatoire même après l’auto-injection
Dépistage pré-administration et contre-indications : conformes à celles de l’infliximab
Efficacité dans la maladie de Behçet : stade d’accumulation de cas en cours3)
Précautions concernant l’administration systémique de corticostéroïdes
Lors de la réduction ou de l’arrêt, de graves poussées inflammatoires oculaires peuvent être déclenchées, aggravant le pronostic visuel. En général, l’administration orale prolongée de corticostéroïdes n’est pas recommandée, mais elle peut être utilisée pendant une très courte période d’environ une semaine en cas de modifications exsudatives marquées de la macula3).
Azathioprine : souvent utilisé à l’étranger. Parfois considéré comme traitement de première intention12)
Injection intravitréenne de triamcinolone acétonide : effet suppressif sur les poussées tant que le médicament est dans le corps vitré. Des injections répétées sont nécessaires, attention aux effets secondaires de cataracte et d’augmentation de la pression intraoculaire.
Interféron alpha-2a : principalement utilisé en Europe. Une efficacité élevée a été rapportée12).
Précautions pour les patients chirurgicaux (directives d’utilisation des inhibiteurs du TNF)9)
Pour la chirurgie intraoculaire peu invasive, l’arrêt des inhibiteurs du TNF n’est pas une indication absolue. Pour la chirurgie extraoculaire ou la chirurgie d’autres organes plus invasive, envisager l’arrêt (demi-vie de l’infliximab environ 8 à 9,5 jours, de l’adalimumab environ 14 jours).
Glaucome secondaire : ajouter des collyres, des médicaments oraux ou des perfusions antiglaucomateux.
Cataracte concomitante : une période sans poussée d’au moins 6 mois est souhaitable. Chirurgie après contrôle de l’inflammation (implantation de lentille intraoculaire possible).
Œdème maculaire cystoïde : injection sous-ténonienne de stéroïdes, renforcement des immunosuppresseurs.
Photocoagulation rétinienne : réalisée pour une vascularite occlusive, mais à ne pas pratiquer à la légère car elle peut déclencher une grave crise inflammatoire oculaire3)
QChez quels patients l'infliximab est-il utilisé ?
A
Il est utilisé dans les cas réfractaires et sévères où les crises oculaires ne sont pas contrôlées par la colchicine ou la ciclosporine. Seuls les médecins spécialistes de la Société japonaise d’ophtalmologie et membres de la Société japonaise d’uvéite, ayant suivi une formation en ligne, peuvent le prescrire9). Après une dose initiale à 0, 2 et 6 semaines, le traitement est poursuivi toutes les 8 semaines. Un dépistage de la tuberculose et de l’hépatite B est obligatoire avant l’administration.
6. Physiopathologie et mécanisme détaillé de la maladie
Le mécanisme de la maladie de Behçet serait dû à une combinaison de prédispositions immunogénétiques et de facteurs environnementaux.
Facteurs génétiques : HLA-B51 est le marqueur génétique le plus fortement associé à la maladie de Behçet, présent chez environ 50 % des patients (contre 15 % dans la population générale)5). HLA-B15, B27, B40, B44, B52, B57 et A26 ont également été identifiés comme des allèles de risque indépendants2).
Mécanisme inflammatoire : Le dysfonctionnement des neutrophiles joue un rôle central, et la surproduction de cytokines, notamment le TNF-α, déclenche la réaction inflammatoire. Dans l’œil, une vascularite occlusive se produit, se manifestant par une augmentation de la perméabilité des capillaires rétiniens (fuite fluorescente en forme de fougère à l’angiographie à la fluorescéine). Pendant la phase aiguë, une infiltration de leucocytes (neutrophiles) et des exsudats blancs dus à l’ischémie se forment.
Caractéristiques de l’inflammation non granulomateuse : Iridocyclite non granulomateuse où les cellules inflammatoires ne forment pas d’agrégats → précipités rétro-cornéens fins. C’est une caractéristique importante pour la différenciation avec les uvéites granulomateuses comme la sarcoïdose et la maladie de Harada3).
Répétition paroxystique de l’inflammation : La répétition des crises oculaires conduit à une atrophie rétinienne et une atrophie optique, entraînant une altération sévère de la fonction visuelle. L’hypopyon, de nature neutrophilique et de consistance fluide, reflète également le mécanisme inflammatoire à prédominance neutrophile.
Dans les cas représentatifs des directives, une évolution favorable sur 3 ans et 6 mois a été rapportée3). L’introduction de l’infliximab réduit considérablement la fréquence des crises oculaires et maintient ou améliore l’acuité visuelle10). Une revue sur l’administration à long terme de l’infliximab pour la maladie de Behçet réfractaire au Japon a également montré une efficacité élevée dans le contrôle des crises et le maintien de l’acuité visuelle11).
Une comparaison des patients des années 1980 et 1990 (Yoshida 2004) a confirmé une tendance à l’atténuation de la sévérité4). La prévalence est passée de 6,2 % en 2002 à 3,9 % en 20095). Parallèlement à la diffusion des agents biologiques, l’incidence des troubles visuels sévères a également diminué.
Les inhibiteurs du TNF (infliximab, adalimumab) ont montré leur efficacité dans plusieurs maladies, y compris la maladie de Behçet, dans les uvéites non infectieuses 12). En particulier chez les enfants, un traitement agressif incluant l’infliximab a permis de maintenir une bonne acuité visuelle1). L’adalimumab est en phase d’accumulation de preuves pour la maladie de Behçet, et les résultats de futures recherches sont attendus 9).
:::danger Avertissement
Cet article vise à fournir des informations médicales et ne constitue pas une instruction pour un diagnostic ou un traitement individuel. Toute décision thérapeutique doit suivre les instructions d’un médecin spécialiste. La posologie et le mode d’administration des médicaments varient selon les conditions individuelles ; veuillez consulter votre médecin traitant pour une prescription réelle.
:::
Casem Azri, Perrine Dusser, Laura Eid, Emmanuel Barreau, Isabelle Kone-Paut, Charlotte Borocco, Caroline Galeotti, Sami Saad, et al. Ocular involvement in pediatric Behçet’s disease: is it different than in adults? (a short case series and mini review). BMC Ophthalmol. 2023;23(1). doi:10.1186/s12886-023-03197-5.
Xin Yao, Xing-Ning Wang, Jian-Ming Lai. Pediatric Behçet’s disease with cardiac valvular lesions: A case-based review. Science Progress. 2023;106(2). doi:10.1177/00368504231173404.
Yoshida A, Kawashima H, Motoyama Y, Shibui H, Kaburaki T, Shimizu K, Ando K, Hijikata K, Izawa Y, Hayashi K, Numaga J, Fujino Y, Masuda K, Araie M.. Comparison of patients with Behçet’s disease in the 1980s and 1990s. Ophthalmology. 2004;111(4):810-815. doi:10.1016/j.ophtha.2003.07.018. PMID:15051217.
Kaburaki T, Namba K, Sonoda KH, Kezuka T, Keino H, Fukuhara T, Kamoi K, Nakai K, Mizuki N, Ohguro N, Ocular Behçet Disease Research Group of Japan.. Behçet’s disease ocular attack score 24: evaluation of ocular disease activity before and after initiation of infliximab. Jpn J Ophthalmol. 2014;58(2):120-130. doi:10.1007/s10384-013-0294-0. PMID:24482146.
INTERNATIONALSTUDYGROUPFORBEHC. Criteria for diagnosis of Behcet’s disease. The Lancet. 1990;335(8697). doi:10.1016/0140-6736(90)92643-v.
Ohno S, Nakamura S, Hori S, Shimakawa M, Kawashima H, Mochizuki M, Sugita S, Ueno S, Yoshizaki K, Inaba G.. Efficacy, safety, and pharmacokinetics of multiple administration of infliximab in Behçet’s disease with refractory uveoretinitis. J Rheumatol. 2004;31(7):1362-1368. PMID:15229958.
Namba K, Goto H, Kaburaki T, et al. A major review: current aspects of ocular Behçet’s disease in Japan. Ocul Immunol Inflamm. 2015;23(Suppl 1):S1-S23. doi:10.3109/09273948.2014.981547.
Pasadhika S, Rosenbaum JT.. Update on the use of systemic biologic agents in the treatment of noninfectious uveitis. Biologics. 2014;8:67-81. doi:10.2147/btt.s41477. PMID:24600203; PMCID:PMC3933243.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.