Der Morbus Behçet (Behçet-Krankheit) ist eine systemische entzündliche Erkrankung unbekannter Ursache, die durch die vier Hauptsymptome orale Aphthen, Augenläsionen, Genitalulzera und Hautläsionen gekennzeichnet ist. Die Entzündung ist akut und vorübergehend, aber die wiederholten Rückfälle sind charakteristisch für die Erkrankung. Sie tritt bei jungen Erwachsenen im Alter von 20 bis 50 Jahren auf, das Geschlechterverhältnis beträgt etwa 1:1. Eine Uveitis tritt bei etwa 70 % der männlichen und 45 % der weiblichen Patienten auf, wobei schwere Fälle häufiger bei jungen Männern vorkommen.
Die Erkrankung tritt häufig entlang der Seidenstraße vom Mittelmeerraum bis nach China und Japan auf. Etwa 50 % der Patienten sind HLA-B51-positiv (im Vergleich zu 15 % der Allgemeinbevölkerung) 5), und HLA-A26 (A*2601) wurde ebenfalls als unabhängiger Risikoallel beschrieben. Es wird angenommen, dass immunogenetische Prädisposition und Umweltfaktoren an der Entstehung beteiligt sind. Früher war die Erkrankung mit einer hohen Erblindungsrate verbunden, aber durch Fortschritte bei Immunsuppressiva und Biologika ist die Erblindungsrate heute gesunken.
Der Morbus Behçet ist als seltene Erkrankung Nr. 56 registriert 8).
Merkmale der Erkrankung
Alter bei Beginn: 20–50 Jahre, junge Erwachsene
Geschlechterverteilung: Verhältnis Männer zu Frauen etwa 1:1. Augenläsionen treten häufiger bei jungen Männern auf und sind schwerwiegender.
Geografische Verteilung: Häufig entlang der Seidenstraße (Mittelmeerraum bis Naher Osten bis Ostasien)
Genetischer Hintergrund: HLA-B51-positiv (etwa 50 % der Patienten), HLA-A26 ebenfalls unabhängiger Risikofaktor 5)
4 Hauptsymptome
Orale Aphthen: schmerzhafte, wiederkehrende Geschwüre. Häufig das erste Symptom
Der Anteil des Morbus Behçet an der Gesamtzahl der Uveitiden ist im Laufe der Jahre rückläufig. In einer epidemiologischen Studie von 2002 betrug er 6,2 % (Rang 3), während er 2009 auf 3,9 % (Rang 6) gesunken war5). Neben einem Rückgang der Patientenzahlen wird auch eine mildere Erkrankung berichtet4).
QWas ist Morbus Behçet für eine Erkrankung?
A
Der Morbus Behçet ist eine systemische entzündliche Erkrankung mit wiederkehrenden Schüben, die durch vier Hauptsymptome gekennzeichnet ist: orale Aphthen, Augenbeteiligung, Genitalulzera und Hautläsionen. Die Ursache ist unbekannt, aber es wird ein autoimmuner Mechanismus vermutet. Die Erkrankung tritt häufig entlang der Seidenstraße auf, und es wird angenommen, dass genetische Faktoren (HLA-B51) und Umweltfaktoren eine Rolle spielen. Die Augenbeteiligung ist besonders wichtig, da sie ohne angemessene Behandlung zur Erblindung führen kann.
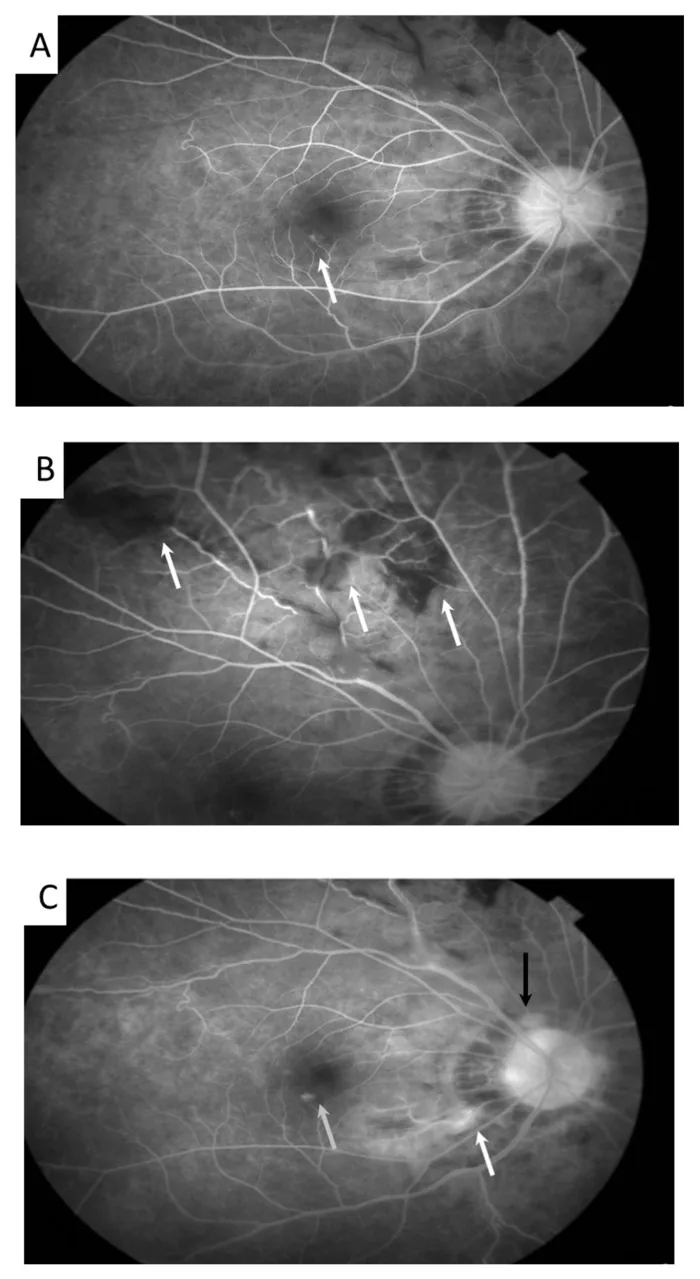
Meng PP, et al. Use of Ultra-Widefield Fluorescein Angiography to Guide the Treatment to Idiopathic Retinal Vasculitis, Aneurysms, and Neuroretinitis-Case Report and Literature Review. Medicina (Kaunas). 2022. Figure 2. PMCID: PMC9611749. License: CC BY.
Im Autofluoreszenzbild sind aneurysmatische Erweiterungen der Netzhautarteriolen (Pfeil) sowie geschlängelte Venen zu erkennen. Dies entspricht den Befunden einer retinalen Vaskulitis, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt wird.
Zu den subjektiven Augensymptomen gehören während eines Entzündungsschubs verschwommenes Sehen, verminderte Sehschärfe und Floater. Die Entzündung tritt häufig in verschiedenen Zeitabständen an einem Auge auf, und etwa 90 % der Fälle entwickeln schließlich eine beidseitige Beteiligung. In schweren Fällen kann die Anfallshäufigkeit mehrmals pro Monat betragen, während sie in leichten Fällen etwa einmal pro Jahr über mehrere Jahre bis zu mehr als zehn Jahren anhält. Eine wiederholte auf die Makula beschränkte Retinitis kann zu irreversiblen Sehverlust führen.
Zu den systemischen Symptomen gehören hauptsächlich orale Aphthen (ca. 90 %), Hautläsionen (ca. 75 %) und Genitalulzera (ca. 50 %). Orale Aphthen treten bevorzugt an der Zunge, der Wangenschleimhaut, den Lippen und dem Zahnfleisch auf und sind schmerzhafte Ulzera mit einem roten Rand. Sie treten bei dieser Erkrankung fast immer auf und sind oft das erste Symptom. Sie heilen innerhalb von 10 Tagen ohne Narbenbildung ab, rezidivieren jedoch.
Zu den Nebensymptomen gehören Arthritis ohne Deformität oder Steifheit, Nebenhodenentzündung, gastrointestinaler Morbus Behçet (ileozäkales Ulkus), vaskulärer Morbus Behçet (Vaskulitis) und neurovaskulärer Morbus Behçet (Meningoenzephalitis). Da die extraokulären Symptome und die Augenmanifestationen nicht immer miteinander korrelieren, ist neben der augenärztlichen Beobachtung auch die Beurteilung des Allgemeinzustands wichtig3).
Die folgenden zwei Augensymptome treten anfallsartig wiederholt auf.
(1) Akute Iridozyklitis, die manchmal mit Hypopyon einhergeht.
(2) Diffuse Glaskörpertrübung, Retinochoroiditis und retinale Vaskulitis.
Früh- und Aktivitätsphase des Augenanfalls: Anfallsartige Augenentzündung. Feine Hornhautendothelbeschläge, Vorderkammerentzündungszellen und Hypopyon. Fibrinausschwitzung in der Vorderkammer tritt normalerweise nicht auf. Es bilden sich keine Iris- oder Kammerwinkelknötchen (nicht granulomatös). Im hinteren Augenabschnitt sind Glaskörpertrübung, retinale Exsudatherde und Blutungen charakteristischerweise relativ schnell (1–2 Wochen) reversibel3).
Spätbefunde3): Chorioretinale Atrophie, Weißlichwerden der Netzhautgefäße, Blässe der Papille → führt zu schwerer Sehverschlechterung.
Das Hypopyon bei Morbus Behçet ist aufgrund der vorwiegend neutrophilen Infiltration dünnflüssig und bildet einen sauberen horizontalen Spiegel. Es ist charakteristisch, dass es sich bei Lageänderung leicht verschiebt, im Gegensatz zum zähflüssigen Hypopyon bei HLA-B27-assoziierter Uveitis3). Die Hornhautendothelbeschläge sind fein und zeigen ein nicht-granulomatöses Bild.
Bei akuter Exazerbation einer retinalen Uveitis treten Glaskörpertrübung, retinale Vaskulitis, weiße retinale Exsudatherde und Blutungen auf. Die weißen Exsudatherde sind auf Leukozyteninfiltration und ischämiebedingte Schwellung der Sehnervenfasern zurückzuführen und verschwinden relativ schnell (innerhalb von etwa einer Woche). Aufgrund der obliterierenden Vaskulitis kann es zu Blutungen ähnlich wie bei einem retinalen Venenastverschluss kommen.
In der Fluoreszenzangiographie zeigt sich häufig auch außerhalb von Augenattacken eine ausgedehnte, starke Leckage aus den Netzhautkapillaren (baumartige Fluoreszenzleckage), die als charakteristisch für diese Erkrankung gilt.
Ein Scoring zur Quantifizierung des Entzündungsgrades jeder einzelnen Augenattacke. Es umfasst sechs Items: Entzündung der Vorderkammer (max. 4 Punkte), Glaskörpertrübung (max. 4 Punkte), periphere Netzhautläsionen (max. 8 Punkte), hintere Pol-Netzhautläsionen (max. 4 Punkte), Fovea-Läsionen (max. 2 Punkte) und Sehnervenläsionen (max. 2 Punkte), mit einer maximalen Gesamtpunktzahl von 24. Es wird zur objektiven Beurteilung des Schweregrads von Attacken und zur Bewertung des Behandlungserfolgs eingesetzt 6).
Darüber hinaus können intraokulare Entzündungen zu Katarakt und sekundärem Glaukom führen, die eine Ursache für Sehbehinderung darstellen.
Hautbefunde: Erythema nodosum, das bevorzugt an den Unterschenkeln auftritt, subkutane Thrombophlebitis, sowie follikulitis- oder akneähnliche Hautausschläge im Gesicht, am Hals und am Rücken. Genitalulzera sind schmerzhafte, scharf begrenzte aphthöse Ulzera, die bei Männern bevorzugt am Skrotum und Penis, bei Frauen an den großen und kleinen Schamlippen auftreten.
Auch bei pädiatrischen Patienten wurden schwere Augensymptome wie hintere Uveitis, Netzhautvaskulitis und Papillitis berichtet 1), wobei die Verzögerung der Diagnose im Durchschnitt 11,3 ± 8,5 Monate betragen kann 1).
QWie unterscheidet man ein Hypopyon bei Morbus Behçet?
A
Das Hypopyon bei Morbus Behçet ist aufgrund seiner neutrophilen Natur „dünnflüssig“, bildet einen sauberen horizontalen Spiegel und verschiebt sich bei Lageänderung. Im Gegensatz dazu ist das Hypopyon bei HLA-B27-assoziierter Uveitis zähflüssiger, bildet keinen Spiegel und erscheint erhaben 3). Dieser Unterschied in der Beschaffenheit kann ein diagnostischer Hinweis sein.
Die Ursache des Morbus Behçet ist unbekannt, aber es wird angenommen, dass äußere Faktoren wie pathogene Mikroorganismen (z. B. Streptokokken) und innere Faktoren wie genetische Veranlagung und Immunanomalien beteiligt sind. Funktionsstörungen der Neutrophilen und Anomalien von Zytokinen wie TNF-α spielen eine zentrale Rolle, und es kommt zu anfallsartigen und wiederkehrenden Entzündungsreaktionen, die vor allem die Mundschleimhaut, die Augen, die Haut und die äußeren Genitalien betreffen.
Risikofaktoren
Beschreibung
HLA-B51
Positiv bei etwa 50 % der Patienten (15 % der Allgemeinbevölkerung). Wichtigster genetischer Marker 5)
HLA-A26
Als unabhängiges Risikoallel berichtet 2)
Region/Ethnie
Häufig entlang der Seidenstraße (Mittelmeerraum bis Naher Osten bis Ostasien)
Geschlecht/Alter
Auftreten im Alter von 20–50 Jahren. Schwere Augenbeteiligung häufiger bei jungen Männern
Bei Kindern machen Fälle etwa 1,6–7,7 % aller Morbus-Behçet-Fälle aus 2), und eine schwerere Augenbeteiligung bei HLA-B51-positiven Fällen wurde berichtet 1).
:::tip Hinweise für den Alltag
Colchicin muss langfristig eingenommen werden, um Augenattacken vorzubeugen. Auch wenn die Augenattacken vorübergehend nachlassen, sollte die Medikation nicht eigenmächtig abgesetzt werden. Zudem ist bekannt, dass eine Photokoagulationstherapie schwere Attacken auslösen kann, daher ist eine gründliche Absprache mit dem behandelnden Arzt wichtig.
:::
Die Diagnose des Morbus Behçet erfolgt gemäß den klinischen Leitlinien der Forschungsgruppe für Morbus Behçet des japanischen Ministeriums für Gesundheit, Arbeit und Soziales (1987, kleine Revision 2016). Für die Diagnose basierend auf Augensymptomen wird die klinische Leitlinie für Augenläsionen bei Morbus Behçet (2012) 3) herangezogen.
Der vollständige Typ ist definiert als das Auftreten aller vier Hauptsymptome im Verlauf, der unvollständige Typ als das Auftreten von drei Hauptsymptomen oder zwei Hauptsymptomen und zwei Nebensymptomen (oder typische Augensymptome plus ein weiteres Hauptsymptom und zwei Nebensymptome).
Fünf Nebensymptome (kleine Revision 2016)8): (1) Arthritis ohne Deformität oder Steifheit, (2) Nebenhodenentzündung, (3) gastrointestinale Läsionen, repräsentiert durch ileozökale Ulzera, (4) Gefäßläsionen, (5) mittelschwere bis schwere zentralnervöse Läsionen
Typ
Kriterien
Vollständiger Typ
Alle 4 Hauptsymptome treten auf
Unvollständiger Typ
3 Hauptsymptome oder 2 Hauptsymptome + 2 Nebensymptome
Sonderform
Intestinaler Typ, vaskulärer Typ, neurologischer Typ (wenn die Kriterien für den vollständigen oder unvollständigen Typ erfüllt sind und spezielle Läsionen vorliegen)
Die internationalen Diagnosekriterien der International Study Group for Behçet’s Disease (ISG-Kriterien, 1990) 7) werden ebenfalls international häufig verwendet.
Hautpathergietest: Erythem- und Pustelbildung nach intrakutaner Nadelstichprovokation (Hilfsdiagnostik)
Bei pädiatrischen Fällen kommt es häufig zu einer verzögerten Diagnose; eine Studie berichtete, dass die Zeit vom Auftreten der Augensymptome bis zur Diagnose durchschnittlich 11,3 ± 8,5 Monate betrug1).
HLA-B27-assoziierte Uveitis: Das Hypopyon ist zähflüssig und unregelmäßig mit leicht erhabenem Zentrum. Hintere Augenabschnitte sind fast nie betroffen3). Pilzendophthalmitis: Bildet Hypopyon und Glaskörpertrübungen, ist jedoch progredient. Diabetische Iritis: Bildet selten ein Hypopyon3).
Colchicin oral (nicht zugelassen): 0,5–1,5 mg/Tag, üblich 1 mg/Tag aufgeteilt in 2 Dosen. Bei etwa 60 % der Patienten zeigt sich eine teilweise Besserung3). Nebenwirkungen: gastrointestinale Symptome wie Durchfall, teratogen. Auch wenn die Augenattacken abklingen, ist eine Langzeittherapie erforderlich, mit regelmäßigen Blutuntersuchungen (Überwachung auf Leber-/Nierenschäden, Granulozytopenie, Rhabdomyolyse).
Step 3: Bei unzureichendem Ansprechen auf Colchicin
Ciclosporin (Neoral® 50 mg): 5 mg/kg/Tag als Richtwert (aufgeteilt in 2 Dosen). Monitoring mit Ziel-Talspiegel von 50–200 ng/mL.
:::caution Wichtige Nebenwirkungen von Ciclosporin
Nierenfunktionsstörungen treten häufig auf. Zudem ist bekannt, dass bei etwa 20 % der Patienten eine Neuro-Behçet-Erkrankung auftritt; daher ist bei Langzeittherapie auf neurologische Symptome zu achten3). In Kombination mit Colchicin besteht ein Risiko für Myopathie.
:::
Zulassung: 2007 für therapierefraktäre Uveitis posterior bei Morbus Behçet zugelassen
Indikation: Therapieresistente oder aufgrund systemischer Nebenwirkungen schwierig mit Immunsuppressiva behandelbare refraktäre retinale Uveitis
Dosierung: 5 mg/kg als intravenöse Infusion über mindestens 2 Stunden. Verabreichung 2 und 6 Wochen nach der ersten Dosis, danach alle 8 Wochen. Infusion über einen 1,2 μm Membranfilter. Nach Woche 6 kann die Infusionszeit verkürzt werden, wenn keine Infusionsreaktion auftritt (durchschnittliche Infusionsrate jedoch nicht mehr als 5 mg/kg pro Stunde).
Primäres und sekundäres Therapieversagen: Primäres Versagen (kein Ansprechen von Anfang an) und sekundäres Versagen (nachlassende Wirkung im Verlauf) sind möglich.
Screening vor der Behandlung (Richtlinien für TNF-Inhibitoren)9):
Röntgen-Thorax, Tuberkulin-Hauttest, bei stark positiver Reaktion QFT/T-SPOT
Alte tuberkulöse Läsionen → Isoniazid oral ab 3 Wochen vor bis 9 Monate nach Beginn von Infliximab
HBs-Antigen, HBs-Antikörper, HBc-Antikörper (Überwachung auf HBV-Reaktivierung)
HBs-Antigen positiv → Konsultation eines Hepatologen erforderlich. Auch bei negativem HBs-Antigen, aber positivem HBs-Antikörper/HBc-Antikörper (frühere Infektion) → regelmäßige HBV-DNA-Messung
Überprüfung auf Hepatitis C, HTLV-1 und HIV-Infektion
Grunderkrankungen: Vorliegen von Herzinsuffizienz, demyelinisierenden Erkrankungen, bösartigen Tumoren
Kontraindikationen9): Infektionen einschließlich aktiver Tuberkulose (atypische mykobakterielle Infektionen, Hepatitis-B-Virus-Infektion), Herzinsuffizienz (NYHA III oder höher), bösartige Tumoren, demyelinisierende Erkrankungen (z. B. Multiple Sklerose)
Nebenwirkungsmonitoring9): Regelmäßige periphere Blutuntersuchungen (Leukozyten, Lymphozyten) und biochemische Tests (einschließlich CRP). Achten Sie auf das Auftreten von Tuberkulose und Pneumocystis-Pneumonie (Röntgen-Thorax, CT, β-D-Glucan). Achten Sie auf die Reaktivierung einer früheren Hepatitis-B-Virus-Infektion (HBV-DNA). Achten Sie auf Infusionsreaktionen (Beobachtung während der Infusion und 2 Stunden danach). Achten Sie auch auf verzögerte Überempfindlichkeitsreaktionen (Muskelschmerzen, Hautausschlag, Fieber, Gelenkschmerzen, die 3 oder mehr Tage nach der Infusion auftreten).
Arzt- und Einrichtungsstandards9): Facharzt der Japanischen Ophthalmologischen Gesellschaft und Mitglied der Japanischen Gesellschaft für Augenentzündung, Abschluss des E-Learning-Kurses dieser Gesellschaft. Die Einrichtung muss bei der Japanischen Gesellschaft für Augenentzündung registriert sein. Erforderlich sind die Fähigkeit zur Behandlung schwerwiegender Nebenwirkungen, die Zusammenarbeit mit einem auf TNF-Hemmer spezialisierten Internisten sowie die Fähigkeit zur Behandlung von Atemwegs-/Infektionserkrankungen.
Repräsentativer Fall: 32-jähriger Mann, HLA-B51-positiver Volltyp. Unter Ciclosporin, Prednisolon und Colchicin unzureichend kontrolliert → Einleitung von Infliximab. In den 12 Monaten vor Beginn 3 Augenattacken auf beiden Augen → nach Beginn keine Attacken im ersten Jahr. Korrigierter Visus rechts 1,2/links 0,7 → Verbesserung auf rechts 1,2/links 0,9. Auch nach 3 Jahren und 6 Monaten blieb der Visus gut3).
Zulassung: 2016 für nichtinfektiöse intermediate, posteriore und Panuveitis zugelassen
Anwendung: Initial 80 mg subkutan → nach 1 Woche 40 mg → danach alle 2 Wochen 40 mg subkutan
Selbstinjektion: Nach ausreichender Schulung und Übung möglich. Auch nach Selbstinjektion ist eine Überwachung der Augen- und Allgemeinbefunde alle 2–3 Monate obligatorisch
Screening vor der Gabe und Kontraindikationen: entsprechend Infliximab
Wirksamkeit bei Morbus Behçet: Stadium der zunehmenden Fallakquise3)
Bei Dosisreduktion oder Absetzen können schwere Augenentzündungsschübe ausgelöst werden, die die Sehprognose verschlechtern. Im Allgemeinen wird eine langfristige orale Steroidtherapie nicht empfohlen, kann aber bei ausgeprägten exsudativen Veränderungen der Makula für einen sehr kurzen Zeitraum von etwa einer Woche eingesetzt werden3).
Azathioprin: Wird im Ausland häufig verwendet. Kann auch als Erstlinientherapie eingesetzt werden12)
Intravitreale Injektion von Triamcinolonacetonid: Wirkt anfallsunterdrückend, solange das Medikament im Glaskörper verbleibt. Wiederholte Injektionen erforderlich; Nebenwirkungen wie Katarakt und erhöhter Augeninnendruck beachten.
Interferon-α-2a: Hauptsächlich in Europa verwendet. Hohe Wirksamkeit berichtet12).
Hinweise für chirurgische Patienten (Leitlinie zur Anwendung von TNF-Inhibitoren)9)
Bei minimalinvasiven intraokularen Eingriffen ist ein Absetzen von TNF-Inhibitoren nicht zwingend erforderlich. Bei extraokularen oder größeren invasiven Eingriffen an anderen Organen sollte ein Absetzen erwogen werden (Infliximab-Halbwertszeit ca. 8–9,5 Tage, Adalimumab ca. 14 Tage).
Begleitkatarakt: Eine anfallsfreie Periode von mindestens 6 Monaten ist wünschenswert. Operation nach Entzündungskontrolle (Intraokularlinsenimplantation möglich).
Zystoides Makulaödem: Sub-Tenon-Steroidinjektion und Verstärkung der Immunsuppression.
Glaskörpertrübung, Blutung, traktive Netzhautablösung: Vitrektomie in Betracht ziehen
Netzhautphotokoagulation: Wird bei okklusiver Vaskulitis durchgeführt, sollte jedoch nicht leichtfertig erfolgen, da sie schwere Augenentzündungsanfälle auslösen kann3)
QBei welchen Patienten wird Infliximab eingesetzt?
A
Es wird bei therapierefraktären, schweren Fällen eingesetzt, bei denen Augenanfälle mit Colchicin oder Ciclosporin nicht kontrolliert werden können. Es darf nur von Ärzten verschrieben werden, die Fachärzte der Japanischen Ophthalmologischen Gesellschaft und Mitglieder der Japanischen Gesellschaft für Augenentzündung sind und ein E-Learning absolviert haben9). Nach der initialen Verabreichung in Woche 0, 2 und 6 erfolgt die Fortsetzung in 8-wöchigen Abständen. Vor der Verabreichung ist ein Screening auf Tuberkulose und Hepatitis B obligatorisch.
6. Pathophysiologie und detaillierter Krankheitsmechanismus
Es wird angenommen, dass der Krankheitsmechanismus des Morbus Behçet auf einer komplexen Beteiligung immunogenetischer Prädisposition und Umweltfaktoren beruht.
Genetische Faktoren: HLA-B51 ist der am stärksten mit Morbus Behçet assoziierte genetische Marker und ist bei etwa 50 % der Patienten positiv (15 % in der Allgemeinbevölkerung)5). HLA-B15, B27, B40, B44, B52, B57 und A26 wurden ebenfalls als unabhängige Risikoallele identifiziert2).
Entzündungsmechanismus: Eine Funktionsstörung der Neutrophilen spielt eine zentrale Rolle, und die Überproduktion von Zytokinen, insbesondere TNF-α, löst Entzündungsreaktionen aus. Im Auge kommt es zu einer okklusiven Vaskulitis, die sich als erhöhte Permeabilität der Netzhautkapillaren (farnkrautartige Fluoreszeinleckage in der Fluoreszenzangiographie) äußert. In der akuten Exazerbationsphase bilden sich weiße Exsudatherde aufgrund von Leukozyteninfiltration (Neutrophile) und Ischämie.
Merkmale der nicht-granulomatösen Entzündung: Nicht-granulomatöse Iridozyklitis, bei der sich die Entzündungszellen nicht zu Klumpen zusammenballen → staubförmige Präzipitate. Dies ist ein wichtiges Unterscheidungsmerkmal zu granulomatösen Uveitiden wie Sarkoidose oder Harada-Krankheit3).
Anfallsartig wiederkehrende Entzündung: Mit wiederholten Augenattacken kommt es zu Netzhautatrophie und Optikusatrophie, was zu schweren Sehstörungen führt. Dass das Hypopyon neutrophilenreich und dünnflüssig ist, spiegelt den neutrophilen-dominierten Entzündungsmechanismus wider.
In repräsentativen Fällen der Leitlinie wurde ein günstiger Verlauf über 3 Jahre und 6 Monate berichtet 3). Durch die Einführung von Infliximab nimmt die Häufigkeit von Augenattacken deutlich ab, und das Sehvermögen bleibt erhalten oder verbessert sich 10). Auch in einer Übersicht zur Langzeitanwendung von Infliximab bei refraktärem Morbus Behçet in Japan wurde eine hohe Wirksamkeit bei der Anfallsunterdrückung und dem Erhalt des Sehvermögens gezeigt 11).
Ein Vergleich von Patienten aus den 1980er und 1990er Jahren (Yoshida 2004) bestätigte einen Trend zu milderen Verläufen 4). Von 2002 bis 2009 sank die Prävalenz von 6,2 % auf 3,9 % 5). Parallel zur Verbreitung von Biologika nahm auch die Häufigkeit schwerer Sehstörungen ab.
TNF-Hemmer (Infliximab, Adalimumab) haben bei mehreren nichtinfektiösen Uveitis-Erkrankungen, einschließlich Morbus Behçet, Wirksamkeit gezeigt 12). Insbesondere bei Kindern wurde durch eine aggressive Behandlung mit Infliximab ein guter Visuserhalt berichtet 1). Für Adalimumab bei Morbus Behçet sammelt sich zunehmend Evidenz an, und zukünftige Forschungsergebnisse werden erwartet 9).
:::danger Haftungsausschluss
Dieser Artikel dient der Bereitstellung medizinischer Informationen und ersetzt keine individuelle Diagnose oder Therapie. Entscheidungen über die Behandlung müssen stets gemäß den Anweisungen eines Facharztes getroffen werden. Dosierung und Anwendung von Medikamenten variieren je nach individuellem Krankheitsbild; wenden Sie sich daher für die tatsächliche Verschreibung an Ihren behandelnden Arzt.
:::
Casem Azri, Perrine Dusser, Laura Eid, Emmanuel Barreau, Isabelle Kone-Paut, Charlotte Borocco, Caroline Galeotti, Sami Saad, et al. Ocular involvement in pediatric Behçet’s disease: is it different than in adults? (a short case series and mini review). BMC Ophthalmol. 2023;23(1). doi:10.1186/s12886-023-03197-5.
Xin Yao, Xing-Ning Wang, Jian-Ming Lai. Pediatric Behçet’s disease with cardiac valvular lesions: A case-based review. Science Progress. 2023;106(2). doi:10.1177/00368504231173404.
Yoshida A, Kawashima H, Motoyama Y, Shibui H, Kaburaki T, Shimizu K, Ando K, Hijikata K, Izawa Y, Hayashi K, Numaga J, Fujino Y, Masuda K, Araie M.. Comparison of patients with Behçet’s disease in the 1980s and 1990s. Ophthalmology. 2004;111(4):810-815. doi:10.1016/j.ophtha.2003.07.018. PMID:15051217.
Kaburaki T, Namba K, Sonoda KH, Kezuka T, Keino H, Fukuhara T, Kamoi K, Nakai K, Mizuki N, Ohguro N, Ocular Behçet Disease Research Group of Japan.. Behçet’s disease ocular attack score 24: evaluation of ocular disease activity before and after initiation of infliximab. Jpn J Ophthalmol. 2014;58(2):120-130. doi:10.1007/s10384-013-0294-0. PMID:24482146.
INTERNATIONALSTUDYGROUPFORBEHC. Criteria for diagnosis of Behcet’s disease. The Lancet. 1990;335(8697). doi:10.1016/0140-6736(90)92643-v.
Ohno S, Nakamura S, Hori S, Shimakawa M, Kawashima H, Mochizuki M, Sugita S, Ueno S, Yoshizaki K, Inaba G.. Efficacy, safety, and pharmacokinetics of multiple administration of infliximab in Behçet’s disease with refractory uveoretinitis. J Rheumatol. 2004;31(7):1362-1368. PMID:15229958.
Namba K, Goto H, Kaburaki T, et al. A major review: current aspects of ocular Behçet’s disease in Japan. Ocul Immunol Inflamm. 2015;23(Suppl 1):S1-S23. doi:10.3109/09273948.2014.981547.
Pasadhika S, Rosenbaum JT.. Update on the use of systemic biologic agents in the treatment of noninfectious uveitis. Biologics. 2014;8:67-81. doi:10.2147/btt.s41477. PMID:24600203; PMCID:PMC3933243.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.