La malattia di Behçet è una malattia infiammatoria sistemica cronica di origine sconosciuta, caratterizzata da quattro sintomi principali: ulcere aftose orali, lesioni oculari, ulcere genitali e manifestazioni cutanee. L’infiammazione è acuta e transitoria, ma la caratteristica distintiva è la ricorrenza delle riacutizzazioni. Insorge in giovani adulti tra i 20 e i 50 anni, con un rapporto maschi-femmine di circa 1:1. L’uveite si verifica in circa il 70% dei pazienti maschi e nel 45% delle pazienti femmine, con casi più gravi nei giovani maschi.
È frequente nelle regioni lungo la Via della Seta, dal Mediterraneo alla Cina e al Giappone. Circa il 50% dei pazienti è positivo per HLA-B51 (rispetto al 15% della popolazione generale)5), e HLA-A26 (A*2601) è stato riportato come allele di rischio indipendente. Si ipotizza che fattori immuno-genetici e ambientali contribuiscano all’insorgenza. Un tempo questa malattia aveva un alto tasso di cecità, ma oggi, grazie ai progressi nei farmaci immunosoppressori e nei farmaci biologici, il tasso di cecità è diminuito.
La malattia di Behçet è registrata come malattia rara designata n. 568).
Caratteristiche della malattia
Età di insorgenza: giovani adulti tra i 20 e i 50 anni
Differenza di genere: rapporto maschi-femmine circa 1:1. Le lesioni oculari sono più gravi nei giovani maschi
Distribuzione geografica: frequente nelle regioni lungo la Via della Seta (dal Mediterraneo al Medio Oriente all’Asia orientale)
Background genetico: positività per HLA-B51 (circa il 50% dei pazienti), HLA-A26 è un fattore di rischio indipendente5)
4 sintomi principali
Ulcera aftosa orale: ulcera ricorrente dolorosa. Spesso è il sintomo iniziale
La proporzione della malattia di Behçet tra tutte le uveiti è in diminuzione nel tempo. Secondo un’indagine epidemiologica del 2002 era del 6,2% (terzo posto), ma nel 2009 è scesa al 3,9% (sesto posto)5). Oltre alla diminuzione del numero di pazienti, è stata riportata anche una riduzione della gravità della malattia4).
QChe tipo di malattia è la malattia di Behçet?
A
La malattia di Behçet è una malattia infiammatoria sistemica recidivante caratterizzata da quattro sintomi principali: ulcere aftose orali, sintomi oculari, ulcere genitali e sintomi cutanei. La causa è sconosciuta, ma si ipotizza un meccanismo autoimmune. È più comune lungo la Via della Seta e si ritiene che siano coinvolti fattori genetici (HLA-B51) e ambientali. Le lesioni oculari sono particolarmente importanti e, se non trattate adeguatamente, possono portare alla cecità.
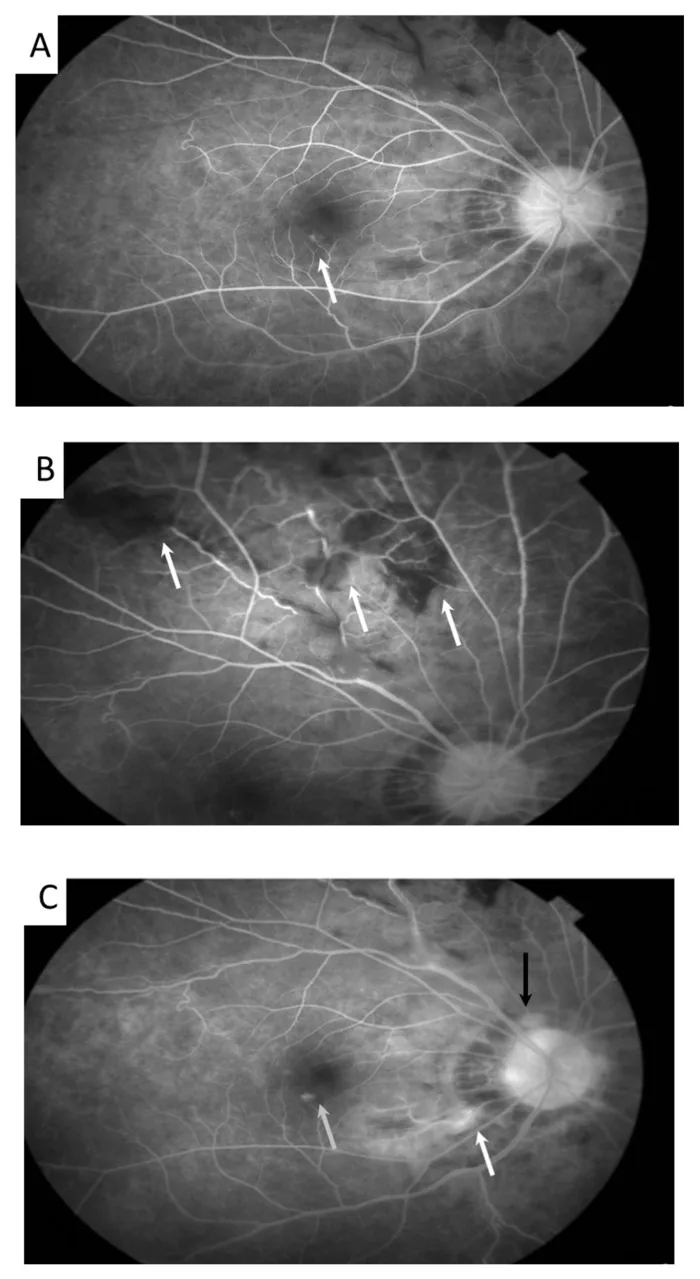
Meng PP, et al. Use of Ultra-Widefield Fluorescein Angiography to Guide the Treatment to Idiopathic Retinal Vasculitis, Aneurysms, and Neuroretinitis-Case Report and Literature Review. Medicina (Kaunas). 2022. Figure 2. PMCID: PMC9611749. License: CC BY.
Nell’immagine di autofluorescenza si osservano dilatazioni aneurismatiche delle arteriole retiniche (freccia) e tortuosità venose. Corrisponde ai reperti di vasculite retinica trattati nella sezione «2. Principali sintomi e reperti clinici».
I sintomi soggettivi oculari includono offuscamento della vista, diminuzione dell’acuità visiva e miodesopsie durante le fasi infiammatorie acute. L’infiammazione spesso si ripresenta in occhi diversi in momenti diversi, e alla fine diventa bilaterale in circa il 90% dei casi. Nei casi gravi, la frequenza degli attacchi può essere di diverse volte al mese, mentre nei casi lievi si verifica circa una volta all’anno per diversi anni o più di un decennio. Può verificarsi retinite ricorrente limitata alla macula, causando una diminuzione irreversibile dell’acuità visiva.
I sintomi sistemici includono principalmente ulcere aftose orali (circa il 90%), sintomi cutanei (circa il 75%) e ulcere genitali (circa il 50%). Le ulcere aftose orali compaiono frequentemente su lingua, mucosa delle guance, labbra e gengive, e sono ulcere dolorose con arrossamento circostante. Sono quasi sempre presenti nella malattia e spesso rappresentano il sintomo iniziale. Guarigione senza cicatrici entro 10 giorni, ma con recidive.
Come sintomi secondari, si possono verificare artrite senza deformità o rigidità, epididimite, malattia di Behçet gastrointestinale (ulcera ileocecale), malattia di Behçet vascolare (vasculite) e malattia di Behçet neurologica (meningoencefalite). Poiché i sintomi extraoculari e i segni oculari non sono sempre correlati, è importante valutare sia le condizioni oculari che lo stato generale del paziente3).
I seguenti due sintomi oculari si ripresentano in modo parossistico.
(1) Irite acuta o iridociclite, talvolta accompagnata da ipopion.
(2) Opacità vitreale diffusa, corioretinite e retinite vascolare.
Segni precoci e di fase attiva dell’attacco oculare: Infiammazione oculare parossistica. Piccoli precipitati cheratici, cellule infiammatorie nella camera anteriore e ipopion. Di solito non si osserva essudazione di fibrina nella camera anteriore. Non si formano noduli iridei o goniosinechie (non granulomatosi). Nel segmento posteriore, è caratteristico che l’opacità vitreale, le placche essudative retiniche e le emorragie scompaiano relativamente rapidamente (1-2 settimane)3).
Segni tardivi3): Atrofia corioretinica, sbiancamento dei vasi retinici, pallore del disco ottico → portano a grave compromissione della funzione visiva.
L’ipopion nella malattia di Behçet è fluido perché è composto principalmente da infiltrazione di neutrofili, formando un bel livello orizzontale. È caratteristico che si sposti facilmente con i cambiamenti di posizione, a differenza dell’ipopion viscoso dell’uveite associata a HLA-B273). I precipitati cheratici sono fini e mostrano segni non granulomatosi.
Durante l’esacerbazione acuta della panuveite, si osservano opacità vitreale, vasculite retinica, placche essudative retiniche bianche ed emorragie. Le placche essudative bianche sono dovute all’infiltrazione di leucociti e al gonfiore delle fibre nervose ottiche causato dall’ischemia, e hanno la caratteristica di scomparire relativamente presto (in circa una settimana). La vasculite occlusiva può causare emorragie simili a quelle dell’occlusione di branca venosa retinica.
Nell’angiografia con fluoresceina, anche in assenza di attacchi oculari, si osserva spesso un’ampia fuoriuscita di fluoresceina dai capillari retinici (fuoriuscita a forma di felce), considerata caratteristica di questa malattia.
Punteggio che quantifica il grado di infiammazione per ogni singolo attacco oculare. Comprende 6 elementi: infiammazione della camera anteriore (max 4 punti), opacità vitreale (max 4 punti), lesioni retiniche periferiche (max 8 punti), lesioni retiniche del polo posteriore (max 4 punti), lesioni foveali (max 2 punti) e lesioni del nervo ottico (max 2 punti), per un totale massimo di 24 punti. Viene utilizzato per la valutazione oggettiva della gravità dell’attacco e per determinare l’efficacia del trattamento6).
Inoltre, l’infiammazione intraoculare può portare a cataratta complicata e glaucoma secondario, causando deficit visivo.
Reperti cutanei: eritema nodoso frequente alle gambe, tromboflebite sottocutanea, rash follicolite o acneiforme su viso, collo e schiena. Le ulcere genitali sono ulcere aftose dolorose con bordi netti, frequenti nello scroto e nel pene negli uomini, e nelle grandi e piccole labbra nelle donne.
Anche nei pazienti pediatrici sono stati riportati sintomi oculari gravi come uveite posteriore, vasculite retinica e papillite1), e in alcuni casi il ritardo diagnostico può arrivare a una media di 11,3±8,5 mesi1).
QCome si distingue l'ipopion nella malattia di Behçet?
A
L’ipopion nella malattia di Behçet è di natura neutrofila, quindi è “fluido” e forma un livello orizzontale netto che si sposta con i cambi di posizione. Al contrario, l’ipopion nell’uveite associata a HLA-B27 è più viscoso, non forma un livello e appare come una massa sollevata3). Questa differenza di consistenza è un indizio diagnostico.
La causa della malattia di Behçet è sconosciuta, ma si ritiene che fattori esterni come i microrganismi patogeni (ad esempio gli streptococchi) e fattori interni come la predisposizione genetica e le anomalie immunitarie siano coinvolti. Le anomalie della funzione dei neutrofili e le anomalie delle citochine, in particolare il TNF-α, svolgono un ruolo centrale, provocando una reazione infiammatoria parossistica e ricorrente che colpisce principalmente la mucosa orale, gli occhi, la pelle e i genitali.
Fattori di rischio
Descrizione
HLA-B51
Positivo in circa il 50% dei pazienti (15% nella popolazione generale). Principale marcatore genetico 5)
HLA-A26
Segnalato come allele di rischio indipendente 2)
Regione/etnia
Alta frequenza nelle regioni lungo la Via della Seta (Mediterraneo, Medio Oriente, Asia orientale)
Sesso/età
Insorgenza tra i 20 e i 50 anni. I giovani uomini presentano spesso lesioni oculari gravi
Nei bambini, i casi rappresentano circa l’1,6-7,7% di tutti i casi di malattia di Behçet2) ed è stato riportato un aggravamento delle lesioni oculari nei casi HLA-B51 positivi1).
:::tip Consigli per la vita quotidiana
La colchicina deve essere assunta a lungo termine per prevenire gli attacchi oculari. Anche se gli attacchi oculari sembrano essersi attenuati, non interrompere l’assunzione del farmaco di propria iniziativa. Inoltre, è noto che il trattamento con fotocoagulazione può talvolta scatenare attacchi violenti, quindi è importante consultare a fondo il medico curante.
:::
La diagnosi della malattia di Behçet si basa sulle linee guida cliniche del gruppo di ricerca sulla malattia di Behçet del Ministero della Salute, del Lavoro e del Welfare (1987, riviste nel 2016). Per la diagnosi basata sui sintomi oculari, fare riferimento alle Linee guida cliniche per le lesioni oculari della malattia di Behçet (2012) 3).
Il tipo completo è definito come la comparsa di tutti e 4 i sintomi principali nel corso della malattia, mentre il tipo incompleto è definito come la comparsa di 3 sintomi principali o di 2 sintomi principali e 2 sintomi secondari (o sintomi oculari tipici più un altro sintomo principale e 2 sintomi secondari).
5 sintomi secondari (revisione 2016)8): (1) artrite senza deformità o rigidità, (2) epididimite, (3) lesioni gastrointestinali rappresentate da ulcere ileocecali, (4) lesioni vascolari, (5) lesioni del sistema nervoso centrale di grado moderato o superiore.
Tipo
Requisiti
Tipo completo
Comparsa di tutti e 4 i sintomi principali
Tipo incompleto
3 sintomi principali o 2 sintomi principali + 2 sintomi secondari
Tipo speciale
Tipo intestinale, vascolare e neurologico (quando soddisfano i criteri di forma completa o incompleta e presentano lesioni speciali)
I criteri diagnostici internazionali dell’International Study Group for Behçet’s Disease (criteri ISG, 1990) 7) sono ampiamente utilizzati a livello internazionale.
Esami del sangue: conta leucocitaria, PCR, VES (marker infiammatori)
Test di iperreattività cutanea (pathergy test): eritema e formazione di pustole dopo puntura intradermica (diagnosi ausiliaria)
Nei pazienti pediatrici si verifica spesso un ritardo diagnostico; è stato riportato che il tempo medio dalla comparsa dei sintomi oculari alla diagnosi è di 11,3±8,5 mesi1).
Uveite associata a HLA-B27: l’ipopion è viscoso, di forma irregolare con una leggera elevazione centrale. Quasi nessuna infiammazione del segmento posteriore3). Endoftalmite fungina: forma ipopion e opacità vitreali, ma progressiva. Irite diabetica: raramente forma un livello liquido3).
Colchicina orale (non coperta da assicurazione): 0,5-1,5 mg/die, di solito 1 mg/die in due dosi divise. Si osserva un miglioramento parziale in circa il 60% dei pazienti3). Effetti collaterali: sintomi gastrointestinali come diarrea, teratogenicità. È necessaria una terapia a lungo termine anche dopo la risoluzione degli attacchi oculari, con regolari esami del sangue (monitoraggio di danno epatico/renale, granulocitopenia, rabdomiolisi).
Ciclosporina (Neoral® 50 mg): circa 5 mg/kg/die (in due dosi divise). Monitorare la concentrazione trough target 50-200 ng/mL.
:::caution Effetti collaterali importanti della ciclosporina
La disfunzione renale si verifica con alta frequenza. Inoltre, è noto che la malattia di Behçet neurologica si manifesta in circa il 20% dei casi, pertanto durante la somministrazione a lungo termine è necessario prestare attenzione ai sintomi neurologici3). In combinazione con colchicina, esiste il rischio di miopatia.
:::
Approvazione assicurativa: Approvato nel 2007 per l’uveite retinica refrattaria dovuta alla malattia di Behçet
Indicazioni: uveite retinica refrattaria resistente ai trattamenti esistenti o quando l’uso di immunosoppressori è difficile a causa di effetti collaterali sistemici
Posologia: 5 mg/kg in infusione endovenosa della durata di almeno 2 ore. Somministrazione a 2 e 6 settimane dopo la prima dose, poi ogni 8 settimane. Infondere attraverso un filtro con pori ≤ 1,2 μm. Dopo la sesta settimana, se non si verificano reazioni all’infusione, è possibile ridurre la durata dell’infusione (ma la velocità media non deve superare 5 mg/kg all’ora)
Non-risposta primaria e secondaria: la non-risposta primaria si verifica quando non si ottiene alcun effetto fin dall’inizio; la non-risposta secondaria si verifica quando l’effetto diminuisce durante il trattamento
Screening pre-trattamento (linee guida per l’uso di farmaci anti-TNF)9):
Radiografia del torace, test cutaneo alla tubercolina; se fortemente positivo, QFT/T-SPOT
Lesioni tubercolari pregresse → somministrazione orale di isoniazide da 3 settimane prima dell’inizio di infliximab fino al 9° mese
HBsAg, HBsAb, HBcAb (monitoraggio della riattivazione di HBV)
HBsAg positivo → consultare obbligatoriamente uno specialista epatologo. Anche se negativo, se HBsAb/HBcAb positivo (infezione pregressa) → misurazione periodica del DNA di HBV
Verifica della presenza di epatite C, HTLV-1 e infezione da HIV
Malattie di base: presenza di insufficienza cardiaca congestizia, malattie demielinizzanti, tumori maligni
Controindicazioni alla somministrazione9): infezioni attive inclusa tubercolosi (infezioni micobatteriche atipiche, infezione da virus dell’epatite B), insufficienza cardiaca congestizia (NYHA classe III o superiore), tumori maligni, malattie demielinizzanti (sclerosi multipla, ecc.)
Monitoraggio degli effetti collaterali9): esami del sangue periferico periodici (globuli bianchi, linfociti), esami biochimici (incluso CRP). Attenzione allo sviluppo di tubercolosi e polmonite da Pneumocystis (radiografia del torace, TC, β-D-glucano). Attenzione alla riattivazione dell’infezione pregressa da virus dell’epatite B (HBV-DNA). Reazioni durante la somministrazione (osservazione durante e per 2 ore dopo). Attenzione anche alle reazioni di ipersensibilità tardiva (dolore muscolare, eruzione cutanea, febbre, artralgia dopo 3 o più giorni dalla somministrazione).
Requisiti del medico e della struttura9): specialista certificato dalla Società Giapponese di Oftalmologia e membro della Società Giapponese di Uveite, completamento dell’e-learning della stessa società. La struttura di introduzione deve essere registrata presso la Società Giapponese di Uveite. È richiesta la capacità di gestire effetti collaterali gravi, la gestione di problemi respiratori/infettivi e la collaborazione con un internista esperto in farmaci anti-TNF.
Caso rappresentativo: uomo di 32 anni, HLA-B51 positivo, forma completa. Controllo difficile con ciclosporina, prednisolone e colchicina → introduzione di infliximab. 3 attacchi oculari in totale in entrambi gli occhi nell’anno precedente l’inizio → nessun attacco nell’anno successivo. L’acuità visiva corretta è migliorata da destra 1.2/sinistra 0.7 a destra 1.2/sinistra 0.9. Buona acuità visiva mantenuta anche dopo 3 anni e 6 mesi3).
Approvazione assicurativa: approvato nel 2016 per uveite intermedia, posteriore e panuveite non infettiva
Dosaggio: 80 mg per via sottocutanea alla prima somministrazione → 40 mg dopo una settimana → successivamente 40 mg ogni due settimane per via sottocutanea
Autoiniezione: possibile dopo un’adeguata formazione e addestramento. Dopo l’autoiniezione, è obbligatorio il monitoraggio dei segni oculari e sistemici ogni 2-3 mesi
Screening pre-trattamento e controindicazioni: come per infliximab
Efficacia nella malattia di Behçet: fase di accumulo di casi in corso3)
Attenzione sulla somministrazione sistemica di steroidi
Durante la riduzione o la sospensione, in alcuni casi si possono scatenare gravi attacchi infiammatori oculari che peggiorano la prognosi visiva. In generale, l’assunzione prolungata di steroidi per via orale non è raccomandata, ma può essere utilizzata per un periodo molto breve di circa una settimana in caso di alterazioni essudative maculari marcate3).
Azatioprina: spesso utilizzata all’estero. Talvolta considerata come prima scelta12)
Iniezione intravitreale di triamcinolone acetonide: effetto soppressivo degli attacchi finché il farmaco è nella cavità vitreale. Richiede iniezioni ripetute; attenzione agli effetti collaterali di cataratta e aumento della pressione intraoculare.
Interferone-alfa-2a: utilizzato principalmente in Europa. È stata riportata un’elevata efficacia12)
Attenzione per i pazienti chirurgici (linee guida per l’uso di farmaci anti-TNF)9)
Per la chirurgia intraoculare mini-invasiva, la sospensione dei farmaci anti-TNF non è un’indicazione assoluta. Per la chirurgia extraoculare o interventi chirurgici maggiori su altri organi, considerare la sospensione (emivita di infliximab circa 8-9,5 giorni, adalimumab circa 14 giorni).
Glaucoma secondario: aggiungere colliri, farmaci orali o endovenosi antiglaucoma.
Cataratta concomitante: è preferibile un periodo senza attacchi di almeno 6 mesi. Chirurgia (con impianto di lente intraoculare) dopo il controllo dell’infiammazione.
Edema maculare cistoide: iniezione di steroidi sottotenoniana e potenziamento dei farmaci immunosoppressori.
Fotocoagulazione retinica: eseguita per la vasculite occlusiva, ma non va effettuata con leggerezza poiché può scatenare gravi attacchi infiammatori oculari3)
QIn quali pazienti viene utilizzato l'infliximab?
A
Viene utilizzato nei casi refrattari e gravi in cui gli attacchi oculari non sono controllabili con colchicina o ciclosporina. Può essere prescritto solo da medici specialisti in oftalmologia riconosciuti dalla Società Giapponese di Oftalmologia e membri della Società Giapponese di Infiammazione Oculare, che abbiano completato l’e-learning9). Dopo la somministrazione iniziale alle settimane 0, 2 e 6, si prosegue con somministrazioni ogni 8 settimane. Prima della somministrazione è obbligatorio lo screening per tubercolosi ed epatite B.
6. Fisiopatologia e meccanismi dettagliati di insorgenza
Si ritiene che la patogenesi della malattia di Behçet sia dovuta a una combinazione di predisposizione immuno-genetica e fattori ambientali.
Fattori genetici: HLA-B51 è il marcatore genetico più fortemente associato alla malattia di Behçet, positivo in circa il 50% dei pazienti (15% nella popolazione generale)5). Anche HLA-B15, B27, B40, B44, B52, B57 e A26 sono stati identificati come alleli di rischio indipendenti2).
Meccanismi infiammatori: l’alterazione funzionale dei neutrofili gioca un ruolo centrale e l’eccessiva produzione di citochine, in particolare TNF-α, scatena la reazione infiammatoria. A livello oculare si verifica una vasculite occlusiva, che si manifesta come aumento della permeabilità dei capillari retinici (perdita di fluoresceina a forma di felce all’angiografia retinica). Durante la fase acuta di esacerbazione si formano infiltrati di leucociti (neutrofili) e placche essudative bianche dovute all’ischemia.
Caratteristiche dell’infiammazione non granulomatosa: iridociclite non granulomatosa in cui le cellule infiammatorie non formano aggregati → KP a polvere fine. Questa è una caratteristica importante per la differenziazione dalle uveiti granulomatose come la sarcoidosi e la malattia di Harada3).
Ripetizione parossistica dell’infiammazione: con il ripetersi degli attacchi oculari, si arriva all’atrofia retinica e all’atrofia del nervo ottico, causando un grave deficit visivo. Il fatto che l’ipopion sia di natura neutrofila e abbia una consistenza fluida riflette il meccanismo infiammatorio prevalentemente neutrofilo.
Nei casi rappresentativi delle linee guida, è stato riportato un buon decorso per 3 anni e 6 mesi3). L’introduzione di infliximab riduce significativamente la frequenza degli attacchi oculari e mantiene o migliora l’acuità visiva10). Anche in una revisione giapponese sulla somministrazione a lungo termine di infliximab per la malattia di Behçet refrattaria, è stata dimostrata un’elevata efficacia nel controllo degli attacchi e nel mantenimento della vista11).
Un confronto tra pazienti degli anni ‘80 e ‘90 (Yoshida 2004) ha confermato una tendenza alla minore gravità4). Dal 2002 al 2009, la prevalenza è diminuita dal 6,2% al 3,9%5). In parallelo con la diffusione dei farmaci biologici, anche l’incidenza di gravi deficit visivi è diminuita.
Gli inibitori del TNF (infliximab, adalimumab) hanno dimostrato efficacia in diverse malattie, tra cui la malattia di Behçet, nelle uveiti non infettive 12). In particolare, anche nei pazienti pediatrici, è stato riportato un buon mantenimento dell’acuità visiva con un trattamento aggressivo che include infliximab1). L’adalimumab è in una fase di accumulo di evidenze per la malattia di Behçet e ci si attendono risultati futuri dalla ricerca 9).
:::danger Dichiarazione di non responsabilità
Questo articolo ha lo scopo di fornire informazioni mediche e non intende indicare procedure diagnostiche o terapeutiche individuali. Per qualsiasi decisione terapeutica, seguire sempre le indicazioni di un medico specialista. Il dosaggio e la modalità di somministrazione dei farmaci variano in base alla condizione individuale; pertanto, per la prescrizione effettiva, consultare il medico curante.
:::
Casem Azri, Perrine Dusser, Laura Eid, Emmanuel Barreau, Isabelle Kone-Paut, Charlotte Borocco, Caroline Galeotti, Sami Saad, et al. Ocular involvement in pediatric Behçet’s disease: is it different than in adults? (a short case series and mini review). BMC Ophthalmol. 2023;23(1). doi:10.1186/s12886-023-03197-5.
Xin Yao, Xing-Ning Wang, Jian-Ming Lai. Pediatric Behçet’s disease with cardiac valvular lesions: A case-based review. Science Progress. 2023;106(2). doi:10.1177/00368504231173404.
Yoshida A, Kawashima H, Motoyama Y, Shibui H, Kaburaki T, Shimizu K, Ando K, Hijikata K, Izawa Y, Hayashi K, Numaga J, Fujino Y, Masuda K, Araie M.. Comparison of patients with Behçet’s disease in the 1980s and 1990s. Ophthalmology. 2004;111(4):810-815. doi:10.1016/j.ophtha.2003.07.018. PMID:15051217.
Kaburaki T, Namba K, Sonoda KH, Kezuka T, Keino H, Fukuhara T, Kamoi K, Nakai K, Mizuki N, Ohguro N, Ocular Behçet Disease Research Group of Japan.. Behçet’s disease ocular attack score 24: evaluation of ocular disease activity before and after initiation of infliximab. Jpn J Ophthalmol. 2014;58(2):120-130. doi:10.1007/s10384-013-0294-0. PMID:24482146.
INTERNATIONALSTUDYGROUPFORBEHC. Criteria for diagnosis of Behcet’s disease. The Lancet. 1990;335(8697). doi:10.1016/0140-6736(90)92643-v.
Ohno S, Nakamura S, Hori S, Shimakawa M, Kawashima H, Mochizuki M, Sugita S, Ueno S, Yoshizaki K, Inaba G.. Efficacy, safety, and pharmacokinetics of multiple administration of infliximab in Behçet’s disease with refractory uveoretinitis. J Rheumatol. 2004;31(7):1362-1368. PMID:15229958.
Namba K, Goto H, Kaburaki T, et al. A major review: current aspects of ocular Behçet’s disease in Japan. Ocul Immunol Inflamm. 2015;23(Suppl 1):S1-S23. doi:10.3109/09273948.2014.981547.
Pasadhika S, Rosenbaum JT.. Update on the use of systemic biologic agents in the treatment of noninfectious uveitis. Biologics. 2014;8:67-81. doi:10.2147/btt.s41477. PMID:24600203; PMCID:PMC3933243.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.