Fase prodromica
Periodo : 1-2 settimane prima della comparsa dei sintomi oculari
Sintomi : Sensazione di formicolio del cuoio capelluto, cefalea (64,7%), acufeni (24,7%), affaticamento (21,1%), sintomi simil-influenzali2)

La malattia di Vogt-Koyanagi-Harada (VKH) è una malattia autoimmune T-cellulare contro la proteina melanica. Nella fase acuta si manifesta con uveite, sintomi di irritazione meningea (cefalea) e perdita dell’udito; con l’infiammazione persistente compaiono manifestazioni cutanee come vitiligine, alopecia e poliosi. Lesioni infiammatorie si verificano in tutti i tessuti contenenti melanociti: occhi, capelli, pelle, orecchio interno e meningi.
Circa la metà dei pazienti presenta sintomi simil-influenzali entro due settimane dall’esordio, suggerendo che un’infezione virale possa fungere da trigger e che la malattia si sviluppi attraverso una reazione crociata immunologica (mimetismo molecolare). Si sospetta il coinvolgimento del virus di Epstein-Barr e del citomegalovirus. La prognosi visiva è generalmente buona, ma in alcuni casi l’infiammazione persistente porta a grave perdita della vista, pertanto sono importanti una diagnosi precoce e un trattamento iniziale.
La malattia di Vogt-Koyanagi-Harada (VKH) è la seconda causa di uveite. In un’indagine epidemiologica del 2002 sono stati riportati 205 casi di VKH (6,7%), e nel 2009 267 casi (7,0%), collocandola al secondo posto dopo la sarcoidosi1). L’età di insorgenza è tra i 20 e i 50 anni, con una leggera prevalenza femminile. L’80% dei pazienti è HLA-DR4 positivo (il 25% della popolazione giapponese normale è HLA-DR4 positivo). La malattia è più comune nelle persone di colore (asiatici, ispanici, nativi americani) e rara nei bianchi. Nello studio FAST, su 93 pazienti VKH, il 71% erano donne, con un’età mediana di 35-38 anni2). È più frequente in Asia orientale, ma si trova anche nelle regioni costiere del Pacifico del Nord e Sud America3).
La malattia è più comune nelle persone di colore (asiatici, ispanici, nativi americani) e rara nei bianchi. Si manifesta preferenzialmente in soggetti HLA-DR4 positivi e nella fascia di età 20-50 anni, con una leggera prevalenza femminile. Si ritiene che sia dovuta a una combinazione di predisposizione genetica (HLA-DRB1*0405) e un fattore scatenante ambientale3).
Fase prodromica
Periodo : 1-2 settimane prima della comparsa dei sintomi oculari
Sintomi : Sensazione di formicolio del cuoio capelluto, cefalea (64,7%), acufeni (24,7%), affaticamento (21,1%), sintomi simil-influenzali2)
Fase acuta (fase di insorgenza dei sintomi oculari)
Improvvisa riduzione della vista : Distacco sieroso retinico bilaterale (57,6% dei VKH acuti)2)
Lieve infiammazione della camera anteriore e opacità vitreali, arrossamento e gonfiore della papilla ottica (circa 70%)
Pieghe coroideali : Nei casi gravi, distacco coroideale e camera anteriore poco profonda
Fase cronica (fase di recidiva)
Infiammazione granulomatosa del segmento anteriore predominante
KP grassi, noduli di Koeppe/Busacca, sinechie posteriori dell’iride
Nel 33,3% dei VKH cronici compare il fondo a cielo serale2)
Fase tardiva (guarigione)
Fondo a cielo serale (sunset glow fundus): perdita di melanina coroidale
Segno di Sugiura: depigmentazione del limbus corneale (circa 1 mese dopo l’esordio)
Sintomi cutanei (circa 20%): vitiligine, alopecia, poliosi (dopo alcuni mesi)
Accumulo di pigmento maculare, dispersione di aree di depigmentazione corioretinica
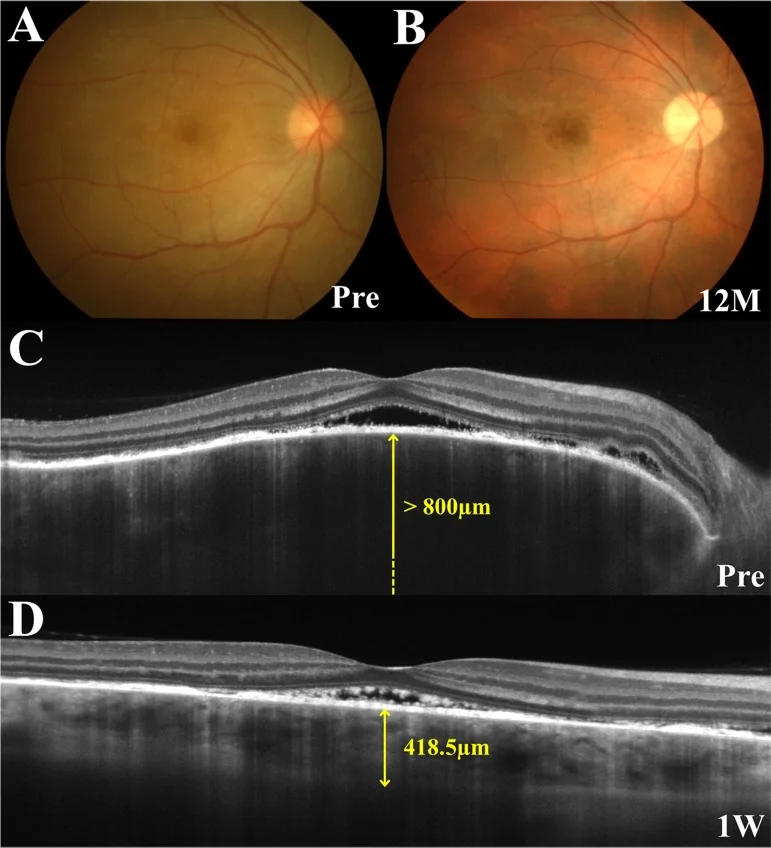
Nella fase prodromica compaiono formicolio del cuoio capelluto, cefalea e acufeni, seguiti 1-2 settimane dopo da un’improvvisa riduzione dell’acuità visiva e offuscamento visivo, che si manifestano simultaneamente o in successione in entrambi gli occhi. All’OCT, la presenza di strutture settate all’interno del distacco retinico acuto è caratteristica.
Sintomi di irritazione meningea (cefalea, rigidità nucale) compaiono precocemente nella maggior parte dei pazienti. Ipoacusia neurosensoriale (poco avvertita ma rilevabile all’esame). I sintomi cutanei (vitiligine, alopecia, poliosi) compaiono alcuni mesi dopo l’esordio e si osservano in circa il 20% dei casi.
In caso di recidiva, il distacco sieroso retinico del segmento posteriore, tipico del primo episodio, è raro; predomina l’infiammazione granulomatosa del segmento anteriore. Si osservano precipitati corneali grassi (KP), noduli di Koeppe e Busacca, e sono frequenti le sinechie posteriori dell’iride. Possono comparire sintomi prodromici come arrossamento oculare e offuscamento visivo.
La reazione autoimmune dei linfociti T CD4-positivi contro le proteine della melanina (famiglia delle tirosinasi: tirosinasi, TRP-1, TRP-2, gp100) è al centro della patologia 3). Gli organi bersaglio sono i melanociti dell’uvea (coroide), del sistema nervoso centrale (meningi), dell’orecchio interno e della pelle; l’infiammazione granulomatosa della coroide è la lesione iniziale principale.
È nota una forte associazione con HLA-DR4 (in particolare DRB10405) come predisposizione genetica 3). È stato riportato anche uno squilibrio di linkage con HLA-DPB10501 3). Esiste l’ipotesi che le infezioni virali (EBV, CMV) inneschino l’autoimmunità attraverso il mimetismo molecolare.
Principali fattori di rischio:
| Tipo | Criteri |
|---|---|
| Malattia di VKH completa | Segni oculari + segni neurologici/uditivi + manifestazioni cutanee, tutti presenti |
| Malattia di VKH incompleta | Segni oculari presenti, ma assenza di segni neurologici/uditivi o manifestazioni cutanee |
| Malattia di VKH probabile | Solo segni oculari (uveite isolata) |
Nelle fasi iniziali non si osservano manifestazioni cutanee, quindi la maggior parte dei casi è di tipo incompleto. Successivamente, quando compaiono le manifestazioni cutanee, diventano di tipo completo. Il distacco sieroso retinico bilaterale precoce è caratteristico e la diagnosi è facile se sono presenti sintomi prodromici come cefalea o sintomi extraoculari come acufeni 1). Nei casi atipici (edema della papilla ottica, unilaterale) l’esame del liquido cerebrospinale è determinante per la diagnosi.
Esame del liquido cerebrospinale: pleiocitosi a predominanza linfocitaria. Persiste fino a 8 settimane. Il più utile per confermare la diagnosi.
Angiografia con fluoresceina (FA): inizialmente ipofluorescenza a chiazze per ritardato riempimento coroidale → fase intermedia: perdita puntiforme (pinpoint leakage) → fase tardiva: accumulo di colorante nell’area di SRD. Iperfluorescenza della papilla ottica (circa 70%) 1).
Angiografia con verde indocianina (ICG): inizialmente ritardato riempimento coroidale a chiazze e perdita di colorante dai vasi coroidali, fase intermedia e tardiva: chiazze ipofluorescenti sparse. È caratteristica anche la sfocatura dei vasi medi e grandi dovuta a disturbo circolatorio coroidale.
OCT: rilevamento e monitoraggio del distacco sieroso retinico. Inizialmente marcato ispessimento coroidale. L’EDI-OCT consente di osservare in dettaglio la sezione trasversale della coroide. All’interno dell’SRD possono essere presenti strutture simil-membranose di fibrina e setti 1).
Ecografia in modalità B: ispessimento coroidale. Utile per la diagnosi differenziale con la sclerite posteriore 1).
HLA di classe II: HLA-DR4 (esame complementare). Positività nell’80% ma specificità bassa (25% di positivi anche nei soggetti sani).
ERG (elettroretinogramma): nella fase cronica riduzione delle ampiezze fotopiche e scotopiche 3).
| Malattia differenziale | Punti di differenziazione |
|---|---|
| Sclerite posteriore | Unilateralità, segno T all’ecografia, dolore ai movimenti oculari |
| Epiteliopatia pigmentaria posteriore multifocale (MPPE) | Unilateralità, distacco sieroso retinico bolloso mobile |
| Epiteliopatia pigmentaria placoida multifocale posteriore acuta (APMPPE) | Fenomeno di inversione all’angiografia con fluoresceina (iperfluorescenza precoce → ipofluorescenza tardiva), giovane età, infezione pregressa |
| Corioretinopatia sierosa centrale (CSCR) | Unilaterale, predominanza maschile, gli steroidi sono un fattore aggravante3) |
| Oftalmia simpatica | Differenziazione tramite anamnesi di trauma oculare perforante o chirurgia intraoculare. Patologicamente considerata la stessa malattia1) |
| Sindrome da essudazione uveale idiopatica | Associazione con nanoftalmo, attenzione alla chiusura d’angolo |
Non è obbligatorio, ma è determinante nei casi atipici di difficile diagnosi (tipo con edema della papilla ottica, caso unilaterale). La pleiocitosi a predominanza linfocitaria persiste fino a 8 settimane, quindi eseguirlo nel momento appropriato dopo l’esordio aumenta l’accuratezza diagnostica. Nei casi tipici con distacco sieroso retinico bilaterale evidente, la diagnosi può spesso essere posta solo sulla base dei reperti clinici.
La terapia con boli di steroidi seguita da riduzione graduale per via orale ad alte dosi è lo standard. Senza trattamento, molti casi vanno incontro a recidive o cronicizzazione, portando a grave deficit visivo. Poiché nella medicina attuale non esiste un indicatore per distinguere i casi di guarigione spontanea da quelli recidivanti/cronici, se possibile si raccomanda la terapia steroidea ad alte dosi.
Esempio di prescrizione rappresentativa delle linee guida1):
Schema di riduzione orale (Prednisone 5 mg compresse):
| Dose | Durata |
|---|---|
| 200 mg/giorno | 2 giorni |
| 150 mg/giorno | 2 giorni |
| 100 mg/giorno | 2 giorni |
| 80 mg/giorno | 2 giorni |
| 60 mg/giorno | 4 giorni |
| 40 mg/giorno | 10 giorni |
| 30 mg/giorno | 2 settimane |
| 20 mg/giorno | 4 settimane |
| 15 mg/giorno | 4 settimane |
| 10 mg/giorno | 4 settimane |
| 5 mg/giorno | 4 settimane |
| 5 mg/giorno (a giorni alterni) | 4 settimane |
La riduzione deve essere lenta e il trattamento deve essere sospeso in un periodo di almeno 6 mesi anche in assenza di recidiva. Con un trattamento inferiore a 6 mesi, il tasso di recidiva è del 58,8%, mentre con 6 mesi o più scende all’11,1% 5). In caso di recidiva, aumentare la dose di steroidi e ridurla gradualmente in un periodo più lungo rispetto alla volta precedente. Con la monoterapia steroidea, il 44% dei pazienti presenta una recidiva e il 59% sviluppa un fundus a tramonto 2).
Dosi per pazienti pediatrici: Nei bambini di età e peso adeguati, si esegue un’infusione endovenosa di metilprednisolone 500 mg/die per 3 giorni (terapia pulsata) o una riduzione orale di prednisolone iniziando con 0,5-1,0 mg/kg/die. La riduzione viene effettuata monitorando gli effetti collaterali.
Ciclosporina (Neoral®): 3 mg/kg/die (per peso 60 kg: 180 mg/die in 2 dosi). È necessario il monitoraggio regolare della concentrazione trough. Consente di ridurre la dose di steroidi. Applicabile nei casi prolungati 1). Attenzione agli effetti collaterali come suscettibilità alle infezioni, disfunzione renale ed epatica.
Metotrexato (MTX): 25 mg/settimana per via orale. Iniziare con 15 mg/settimana per 2 settimane, poi aumentare 2).
Micofenolato mofetile (MMF): 1,5 g due volte al giorno per via orale. Iniziare con 500 mg due volte al giorno e aumentare gradualmente 2).
Lo studio FAST (NCT01829295) è un RCT sulle uveiti non infettive (sottoanalisi di 93 casi di VKH su 216 pazienti). 49 pazienti sono stati randomizzati a MTX e 44 a MMF, con prednisone 1 mg/kg/die (max 60 mg/die) all’inizio e successiva riduzione (obiettivo ≤ 7,5 mg/die a 6 mesi).
Endpoint primario a 6 mesi (sottoanalisi VKH) :
| Indicatore | Gruppo MTX | Gruppo MMF | Valore P |
|---|---|---|---|
| Tasso di successo del trattamento | 80,4% (37/46 casi) | 64,1% (25/39 casi) | 0,10 |
| Riduzione dello spessore retinico foveale | −62,5 μm | −4,0 μm | 0,003 |
| Tasso di scomparsa del SRD | 86,3% | 64,1% | 0,02 |
| Miglioramento della vista | Equivalente | Equivalente | 0,78 |
Il tasso di successo complessivo del trattamento è stato del 74,7% (62/85 casi). Nella VKH acuta, MTX è risultato superiore nella riduzione dello spessore retinico centrale (CST) e nella scomparsa del distacco sieroso retinico (SRD). A 12 mesi, il 91,3% del gruppo MMF manteneva il successo terapeutico e circa la metà (MTX 50,0%, MMF 56,5%) aveva raggiunto la sospensione completa del prednisone 2). Dopo il fallimento del primo trattamento, il passaggio da MMF a MTX ha avuto successo nell’81,8% dei casi 2). Con l’uso iniziale di MMF in combinazione, il 93% dei pazienti con VKH acuta ha mantenuto un visus di 20/20 e non sono state segnalate recidive né formazione di fundus sunset glow 2).
Nei pazienti in cui la somministrazione sistemica di steroidi è difficile (anziani, donne in gravidanza, diabetici, storia di malattie mentali), prendere in considerazione l’iniezione sottotenoniana posteriore di triamcinolone acetonide 1).
Cataratta concomitante: L’uso massiccio di steroidi porta a un’alta incidenza di cataratta steroidea. I casi in remissione completa presentano un rischio simile a quello della chirurgia standard. L’impianto di IOL non è problematico. Se in futuro potrebbe essere necessario un intervento filtrante, scegliere un’incisione corneale preservando la congiuntiva superiore.
Glaucoma secondario / glaucoma steroideo: Utilizzare colliri ipotensivi (beta-bloccanti, analoghi delle PG, inibitori dell’anidrasi carbonica) → CAI orali → infusione di D-mannitolo in quest’ordine. Eseguire una trabeculotomia (efficace nel glaucoma steroideo), se insufficiente, una trabeculectomia.
Anche in assenza di recidiva, si raccomanda di ridurre gradualmente la dose nell’arco di almeno 6 mesi. La sospensione in meno di 6 mesi comporta un tasso di recidiva elevato di circa il 58,8%, mentre la prosecuzione oltre i 6 mesi lo riduce all’11,1% 5). In caso di recidiva, ridurre la dose più lentamente rispetto alla volta precedente e considerare l’aggiunta di un immunosoppressore.
La ciclosporina (Neoral® 3 mg/kg/die) è utilizzata come farmaco risparmiatore di steroidi nei casi persistenti 1). Lo studio FAST ha confermato l’efficacia del metotrexato (MTX 25 mg/settimana) e del micofenolato mofetile (MMF 1,5 g × 2 volte/die). Nella VKH acuta, il MTX ha mostrato una tendenza superiore nella riduzione dello spessore retinico foveale e nel tasso di scomparsa del distacco sieroso retinico 2).
La reazione autoimmune cellulo-mediata dei linfociti T CD4+ contro le proteine della melanina (tirosinasi, TRP-1, TRP-2, gp100) è il meccanismo centrale 3). I bersagli sono i melanociti dell’uvea (coroide), del sistema nervoso centrale (meningi), dell’orecchio interno e della pelle; l’infiammazione granulomatosa della coroide è la lesione iniziale principale.
Come predisposizione genetica è nota una forte associazione con HLA-DR4 (in particolare DRB10405) 3), ed è stato riportato anche uno squilibrio di linkage con HLA-DPB10501 3). Circa la metà dei pazienti presenta sintomi simil-influenzali entro due settimane dall’esordio, suggerendo che un’infezione virale (EBV, CMV) possa scatenare l’autoimmunità attraverso il mimetismo molecolare.
Interpretazione dei reperti di imaging: All’ICG si osservano offuscamento dei vasi medi e grandi e macchie ipofluorescenti dovuti a disturbi circolatori coroideali. L’ispessimento coroideale marcato precoce all’EDI-OCT riflette un edema stromale coroideale. La perdita di melanina nella fase cronica porta al fondo a tramonto 3).
Circa il 25% dei casi presenta infiammazione persistente nonostante la terapia con boli di steroidi; la persistenza dell’infiammazione coroideale porta a un’atrofia retinocoroideale progressiva e a grave compromissione visiva in alcuni pazienti. Spesso permangono sintomi soggettivi come lieve metamorfopsia o anomalie della visione dei colori.
Significato clinico dello studio FAST 2): Primo RCT di confronto diretto tra MTX e MMF nell’uveite non infettiva. I 93 casi di VKH costituivano il sottogruppo più ampio dello studio, dimostrando l’efficacia dell’introduzione precoce di immunosoppressori nella VKH acuta. Sono attesi ulteriori studi di follow-up.
Importanza dell’introduzione precoce di immunosoppressori 2): Con la sola terapia steroidea, il 44% dei pazienti presenta recidive e il 59% sviluppa un fondo a tramonto. La combinazione precoce di steroidi e antimetaboliti può ridurre il tasso di recidiva e l’incidenza del fondo a tramonto. Il concetto di “finestra terapeutica” — l’inizio precoce degli steroidi all’esordio della malattia previene la progressione verso la fase cronica ricorrente e riduce la necessità di terapia immunosoppressiva a lungo termine — sta guadagnando popolarità.
Evoluzione della diagnostica per immagini3): La valutazione quantitativa dello spessore coroideale mediante EDI-OCT e la rilevazione della coroidite occulta mediante ICG sono utilizzate per il monitoraggio terapeutico. La misurazione quantitativa dei foci coroideali iperriflettenti (HCF) mediante en face OCT potrebbe rappresentare un biomarcatore dell’attività della malattia di VKH.
Tendenze nei criteri diagnostici rivisti2): È stata proposta una classificazione precoce/tardiva che incorpora OCT, FA e ICG, con l’obiettivo di migliorare l’accuratezza diagnostica. Gli attuali criteri rivisti del 2001 presentano limiti nella rilevazione della VKH precoce.