前駆期
時期:眼症状発現の1〜2週前
症状:頭髪のピリピリ感、頭痛(64.7%)、耳鳴り(24.7%)、倦怠感(21.1%)、感冒様症状2)

Vogt-小柳-原田病(Vogt-Koyanagi-Harada disease; VKH病)は、メラニン蛋白に対するT細胞性自己免疫疾患である。発症期にぶどう膜炎・頭痛など髄膜刺激症状・難聴を示し、炎症の遷延により白斑・脱毛・白毛などの皮膚症状を呈する全身疾患である。色素細胞(メラノサイト)が分布する全身各所——眼・毛髪・皮膚・内耳・髄膜——に炎症性病変が出現する。
発症前2週間以内に約半数で感冒様症状がみられることから、ウイルス感染が引き金となり免疫学的交差反応(molecular mimicry)により発症する可能性が推測されている。Epstein-Barrウイルスやサイトメガロウイルスなどの関与が示唆される。概して視力予後は良好であるが、炎症の遷延により高度な視力障害に至る症例もあり、早期診断および初期治療が重要である。
ぶどう膜炎の原因疾患として第2位を占める。2002年の疫学調査ではVKH 205例(6.7%)、2009年調査では267例(7.0%)が報告されており、いずれもサルコイドーシスに次ぐ第2位である1)。好発年齢は20〜50歳代で、やや女性に多い。HLA-DR4陽性が患者の80%にみられ(日本人正常人の25%がHLA-DR4陽性)、有色人種(アジア人・ヒスパニック・ネイティブアメリカン)に多く、白人にはまれである。FAST試験に登録されたVKH患者93例のうち女性が71%、年齢中央値35〜38歳であった2)。東アジアに最も多いが、北米や南米の太平洋側の地域にもみられる3)。
有色人種(アジア人・ヒスパニック・ネイティブアメリカン)に多く、白人にはまれである。HLA-DR4陽性者や20〜50歳代にやや女性優位で好発する。遺伝的素因(HLA-DRB1*0405)と何らかの環境的トリガーが組み合わさって発症すると考えられている3)。
前駆期
時期:眼症状発現の1〜2週前
症状:頭髪のピリピリ感、頭痛(64.7%)、耳鳴り(24.7%)、倦怠感(21.1%)、感冒様症状2)
急性期(眼症状発症期)
慢性期(再発期)
前眼部の肉芽腫性炎症が主体
豚脂様KP・Koeppe/Busacca結節、虹彩後癒着
慢性VKHの33.3%に夕焼け状眼底が出現2)
晩期(回復期)
夕焼け状眼底(sunset glow fundus):脈絡膜メラニン色素の脱失
Sugiura徴候:角膜輪部の脱色素(発症後約1か月)
皮膚症状(約20%):白斑・脱毛・白毛(数か月後)
黄斑部色素集積、網脈絡膜脱色素斑の散在
前駆期には頭髪のピリピリ感・頭痛・耳鳴りが出現し、その1〜2週後に急激な視力低下・霧視が両眼同時または前後して発症する。OCT上、急性期の網膜剝離内部には隔壁構造が観察されることが特徴的である。
髄膜刺激症状(頭痛・項部硬直)がほとんどの症例で発症早期に出現する。感音難聴(自覚は少ないが検査で判明)。皮膚症状(白斑・脱毛・白毛)は発症から数か月後に出現し、全体の約20%にみられる。
再発時には初発時のような後眼部の漿液性網膜剝離は少なく、前眼部の肉芽腫性炎症が主体となる。豚脂様角膜後面沈着物(KP)・Koeppe結節・Busacca結節がみられ、虹彩後癒着を生じることが多い。前駆症状として眼の充血・かすみが出現することがある。
メラニン蛋白(チロシナーゼファミリー:チロシナーゼ・TRP-1・TRP-2・gp100)に対するCD4陽性T細胞性自己免疫反応が病態の中核である3)。標的臓器はぶどう膜(脈絡膜)・中枢神経系(髄膜)・内耳・皮膚のメラノサイトであり、脈絡膜の肉芽腫性炎症が初期主病変となる。
遺伝的素因としてHLA-DR4(特にDRB1*0405)との強い関連が知られる3)。HLA-DPB1*0501との連鎖不平衡も報告されている3)。ウイルス感染(EBV・CMV)が分子模倣により自己免疫を誘発するとの仮説がある。
主なリスク因子:
| 型 | 基準 |
|---|---|
| 完全型(Complete VKH disease) | 眼所見+神経学的所見・聴覚所見+皮膚症状の全てを充足 |
| 不全型(Incomplete VKH disease) | 眼所見あり、神経学的所見・聴覚所見または皮膚症状のいずれかを欠く |
| 疑い(Probable VKH disease) | 眼所見のみ(孤立性ぶどう膜炎) |
発症早期には皮膚所見がみられないためほとんどが不全型である。後に皮膚所見が生じると完全型となる。発症早期の両眼性漿液性網膜剝離は特徴的であり、頭痛などの前駆症状や耳鳴などの眼外症状があれば診断は容易である1)。非典型例(視神経乳頭浮腫型・片眼例)では髄液検査が診断の決め手となる。
髄液検査:リンパ球主体の細胞増多。8週間まで持続。診断確定に最も有用。
蛍光眼底造影(FA):初期に脈絡膜充盈遅延を示す斑状低蛍光 → 中期に顆粒状蛍光漏出(pinpoint leakage)→ 後期にSRD部位の色素貯留。視神経乳頭の過蛍光(約70%)1)。
ICG造影:初期に斑状脈絡膜充盈遅延・脈絡膜血管からの色素漏出、中期〜後期に散在性低蛍光斑。脈絡膜循環障害に伴う中大血管の不鮮明化も特徴的である。
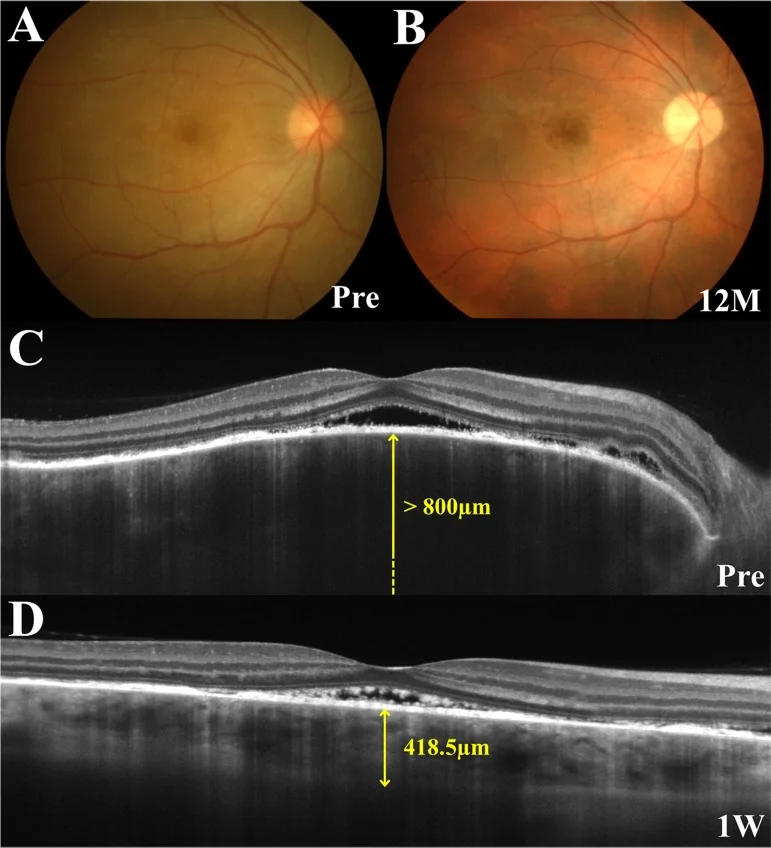
OCT:漿液性網膜剝離の検出・モニタリング。初期に著明な脈絡膜肥厚。EDI-OCTで脈絡膜断面を詳細に観察できる。SRD内にフィブリン膜様構造物・隔壁構造を認めることがある1)。
超音波Bモード:脈絡膜肥厚所見。後部強膜炎との鑑別に有用1)。
HLA class II:HLA-DR4(補助検査)。陽性率80%だが特異性は低い(正常人でも25%陽性)。
ERG(電気網膜図):慢性期に光覚・暗順応の振幅が減少する3)。
| 鑑別疾患 | 鑑別ポイント |
|---|---|
| 後部強膜炎 | 片眼性、超音波でのTサイン、眼球運動痛・疼痛 |
| 多発性後極部色素上皮症(MPPE) | 片眼性、移動性胞状漿液性網膜剝離 |
| 急性後部多発性斑状色素上皮症(APMPPE) | FA逆転現象(早期高蛍光→後期低蛍光)、若年者・先行感染 |
| 中心性漿液性脈絡網膜症(CSCR) | 片眼性、男性優位、ステロイドが増悪因子3) |
| 交感性眼炎 | 穿孔性眼外傷・内眼手術の既往から鑑別。病態的には同一疾患と考えられている1) |
| 特発性ぶどう膜渗出症候群 | ナノフタルモスとの関連、隅角閉塞に注意 |
必須ではないが、診断が困難な非典型例(視神経乳頭浮腫型・片眼例)では決め手となる。リンパ球主体の細胞増多が8週間まで持続するため、発症後の適切な時期に実施することが診断精度を高める。両眼性漿液性網膜剝離が明らかな典型例では、臨床所見のみで診断できることが多い。
ステロイドパルス療法 → 経口大量漸減が標準である。治療を行わずに経過をみた場合、再発・遷延を生じ高度の視力障害に結びつく症例が多い。現在の医学では自然治癒する症例と再発・遷延例を区別する指標がないため、可能であればステロイド大量投与が推奨される。
ガイドライン代表症例処方例1):
経口漸減スケジュール(プレドニン錠5mg):
| 投与量 | 期間 |
|---|---|
| 200mg/日 | 2日間 |
| 150mg/日 | 2日間 |
| 100mg/日 | 2日間 |
| 80mg/日 | 2日間 |
| 60mg/日 | 4日間 |
| 40mg/日 | 10日間 |
| 30mg/日 | 2週間 |
| 20mg/日 | 4週間 |
| 15mg/日 | 4週間 |
| 10mg/日 | 4週間 |
| 5mg/日 | 4週間 |
| 5mg/日(隔日) | 4週間 |
減量はゆっくり行い、再発なくても6か月以上かけて中止する。6か月未満の治療では再発率58.8%、6か月以上では11.1%に低下する5)。再発した場合はステロイドを増量し、前回よりも時間をかけて漸減する。ステロイド単独療法では44%が再発し、59%にsunset glow fundusが発現する2)。
小児患者の治療量:一定以上の年齢・体重のある患児ではメチルプレドニゾロン500mg/日の点滴を3日間施行(パルス療法)か、プレドニゾロン0.5〜1.0mg/kg/日からの内服漸減が行われる。副作用に注意しながら漸減していく。
シクロスポリン(ネオーラル®):3mg/kg/日(体重60kgで180mg/日 分2)。トラフ濃度定期測定が必要。ステロイドの投与量減量が期待できる。遷延例に適用する1)。易感染性・腎機能障害・肝機能障害などの副作用に注意する。
メトトレキサート(MTX):25mg/週 経口投与。初回15mg/週で2週間開始後に増量2)。
ミコフェノール酸モフェチル(MMF):1.5g 1日2回経口投与。初回500mg 1日2回から開始し漸増2)。
FAST試験(NCT01829295)は非感染性ぶどう膜炎対象のRCT(216例中VKH 93例のサブ解析)である。MTX 49例対MMF 44例に無作為割付し、プレドニゾン1mg/kg/日(最大60mg/日)で開始し漸減した(6か月時点で7.5mg/日以下を目標)。
6か月主要アウトカム(VKHサブ解析):
| 指標 | MTX群 | MMF群 | P値 |
|---|---|---|---|
| 治療成功率 | 80.4%(37/46例) | 64.1%(25/39例) | 0.10 |
| 中心窩網膜厚減少 | −62.5μm | −4.0μm | 0.003 |
| SRD消失率 | 86.3% | 64.1% | 0.02 |
| 視力改善 | 同等 | 同等 | 0.78 |
全体の治療成功率は74.7%(62/85例)。急性VKHではMTXがCST減少およびSRD消失で優位であった。12か月時点でMMF群の91.3%が治療成功を継続し、約半数(MTX 50.0%、MMF 56.5%)がプレドニゾン完全離脱を達成した2)。初回治療失敗後のMMF→MTX切替では81.8%が成功した2)。MMF初期併用では急性VKHの93%が20/20を維持し、全例で再発・sunset glow fundus形成なしとの報告がある2)。
ステロイド全身投与困難例(高齢者・妊婦・糖尿病・精神疾患既往)ではトリアムシノロンアセトニド後部テノン囊下注射を検討する1)。
併発白内障:ステロイド大量使用によりステロイド白内障が高率に出現する。完全寛解例は通常の手術と同等リスク。IOL挿入も問題ない。後に濾過手術の可能性がある場合は上方結膜温存・角膜切開を選択する。
続発緑内障・ステロイド緑内障:降圧点眼(β遮断薬・PG製剤・炭酸脱水酵素阻害薬)→ CAI内服 → D-マンニトール点滴の順に用いる。線維柱帯切開術(ステロイド緑内障に有効)、不十分なら線維柱帯切除術を行う。
再発なくても6か月以上かけてゆっくり漸減することが推奨される。6か月未満での中止は再発率が約58.8%と高く、6か月以上継続では11.1%に低下する5)。再発時は前回より時間をかけて漸減し、免疫抑制薬の追加も検討する。
シクロスポリン(ネオーラル® 3mg/kg/日)はステロイド節減薬として遷延例に使用される1)。FAST試験ではメトトレキサート(MTX 25mg/週)とミコフェノール酸モフェチル(MMF 1.5g×2回/日)の有効性が確認された。急性VKHではMTXが中心窩網膜厚減少とSRD消失率で優位な傾向を示した2)。
メラニン蛋白(チロシナーゼ・TRP-1・TRP-2・gp100)に対するCD4陽性T細胞性自己免疫反応が中心的病態である3)。標的はぶどう膜(脈絡膜)・中枢神経(髄膜)・内耳・皮膚のメラノサイトであり、脈絡膜の肉芽腫性炎症が初期主病変となる。
遺伝的素因としてHLA-DR4(特にDRB1*0405)との強い関連が知られ3)、HLA-DPB1*0501との連鎖不平衡も報告されている3)。発症前2週間以内に約半数で感冒様症状がみられることから、ウイルス感染(EBV・CMV)が分子模倣により自己免疫を誘発するとの仮説がある。
画像所見の解釈:ICGでは脈絡膜循環障害に伴う中大血管の不鮮明化・低蛍光斑が観察される。EDI-OCTによる初期の著明な脈絡膜肥厚は脈絡膜間質浮腫を反映する。慢性期のメラニン脱失により夕焼け状眼底が形成される3)。
ステロイドパルス療法を行っても炎症遷延例は約25%にみられ、脈絡膜での炎症の持続により徐々に網脈絡膜萎縮が広がり高度の視力障害に至る症例がある。わずかな歪視や色覚異常などの自覚症状が残ることも多い。
FAST試験の臨床的意義2):非感染性ぶどう膜炎における初のMTX対MMF直接比較RCT。VKH 93例は試験内最大サブグループであり、急性VKHへの早期免疫抑制薬導入の有効性が示された。今後さらなる追跡研究が期待される。
早期免疫抑制薬導入の重要性2):ステロイド単独療法では44%が再発し59%にsunset glow fundusが発現する。早期のステロイド+代謝拮抗薬併用が再発率・sunset glow fundus発現率を低下させる可能性がある。「治療のtherapeutic window」——発症早期のステロイド開始が慢性再発期への進行を予防し、長期免疫抑制療法の必要性を減らすとの考え方が広まりつつある。
画像診断の進化3):EDI-OCTによる脈絡膜肥厚の定量評価、ICGによる潜在性脈絡膜炎の検出が治療モニタリングに活用される。Hyperreflective choroidal foci(HCF)のen face OCTによる定量測定がVKH活動性評価のバイオマーカーとなる可能性がある。
改訂診断基準の動向2):OCT・FA・ICGを取り入れた早期/晩期の分類が提案されており、診断精度向上が期待される。現在の2001年改訂基準では早期VKHの検出に限界があるとの指摘がある。