Fase prodrômica
Período: 1-2 semanas antes do início dos sintomas oculares
Sintomas: Sensação de formigamento no couro cabeludo, dor de cabeça (64,7%), zumbido (24,7%), fadiga (21,1%), sintomas gripais 2)

A doença de Vogt-Koyanagi-Harada (Vogt-Koyanagi-Harada disease; VKH) é uma doença autoimune mediada por células T contra a proteína melanina. Na fase de início, apresenta uveíte, cefaleia, sintomas de irritação meníngea e perda auditiva, e com a inflamação prolongada, surgem sintomas cutâneos como vitiligo, queda de cabelo e cabelos brancos. Lesões inflamatórias aparecem em todos os locais onde os melanócitos estão distribuídos - olhos, cabelo, pele, ouvido interno e meninges.
Como cerca de metade dos pacientes apresenta sintomas gripais dentro de 2 semanas antes do início, suspeita-se que uma infecção viral possa ser o gatilho, levando a uma reação cruzada imunológica (mimetismo molecular). Suspeita-se do envolvimento do vírus Epstein-Barr e do citomegalovírus. Em geral, o prognóstico da acuidade visual é bom, mas em alguns casos, a inflamação prolongada pode levar a deficiência visual grave, portanto, o diagnóstico precoce e o tratamento inicial são importantes.
A VKH ocupa o segundo lugar como causa de uveíte. Em um levantamento epidemiológico de 2002, foram relatados 205 casos de VKH (6,7%) e, em 2009, 267 casos (7,0%), sendo a segunda causa mais comum após a sarcoidose 1). A idade de início é geralmente entre 20 e 50 anos, com leve predominância feminina. Cerca de 80% dos pacientes são HLA-DR4 positivos (25% dos japoneses normais são HLA-DR4 positivos), é mais comum em pessoas de cor (asiáticos, hispânicos, nativos americanos) e raro em brancos. Dos 93 pacientes com VKH registrados no estudo FAST, 71% eram mulheres, com idade mediana de 35-38 anos 2). É mais comum no Leste Asiático, mas também é encontrado em regiões do Pacífico na América do Norte e do Sul 3).
Ocorre mais frequentemente em pessoas de cor (asiáticos, hispânicos, nativos americanos) e é raro em brancos. Ocorre frequentemente em indivíduos HLA-DR4 positivos e na faixa etária de 20-50 anos, com ligeiro predomínio feminino. Acredita-se que a doença ocorra devido a uma combinação de predisposição genética (HLA-DRB1*0405) e um gatilho ambiental específico 3).
Fase prodrômica
Período: 1-2 semanas antes do início dos sintomas oculares
Sintomas: Sensação de formigamento no couro cabeludo, dor de cabeça (64,7%), zumbido (24,7%), fadiga (21,1%), sintomas gripais 2)
Fase aguda (fase de início dos sintomas oculares)
Queda abrupta da visão: Descolamento seroso bilateral da retina (57,6% dos VKH agudos) 2)
Inflamação leve da câmara anterior e opacidade vítrea, rubor e edema do disco óptico (cerca de 70%)
Pregas coroidais: Em casos graves, descolamento da coroide e câmara anterior rasa
Fase crônica (fase de recidiva)
Inflamação granulomatosa do segmento anterior é a principal característica
KP gorduroso, nódulos de Koeppe/Busacca, sinéquias posteriores da íris
Em 33,3% dos casos de VKH crônico, surge fundo de olho em pôr do sol 2)
Fase tardia (fase de recuperação)
Fundo de olho em pôr do sol (sunset glow fundus): perda de melanina da coroide
Sinal de Sugiura: despigmentação do limbo corneano (cerca de 1 mês após o início)
Sintomas cutâneos (cerca de 20%): vitiligo, queda de cabelo, cabelos brancos (após alguns meses)
Acúmulo de pigmento macular, manchas dispersas de despigmentação retino-coroidal
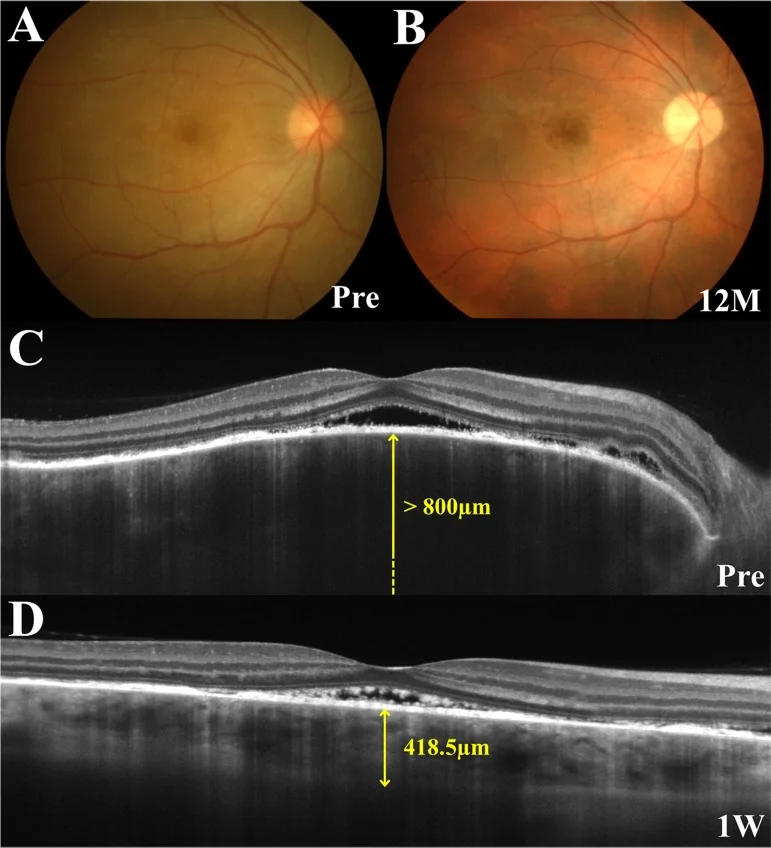
Na fase prodrômica, ocorrem formigamento no couro cabeludo, dor de cabeça e zumbido, e 1-2 semanas depois, diminuição aguda da visão e visão turva em ambos os olhos simultânea ou sequencialmente. Na OCT, a fase aguda é caracterizada por estruturas septadas dentro do descolamento de retina.
Sintomas de irritação meníngea (dor de cabeça, rigidez de nuca) aparecem precocemente na maioria dos casos. Perda auditiva neurossensorial (pouco relatada, mas detectada em exames). Sintomas cutâneos (vitiligo, queda de cabelo, cabelos brancos) surgem meses após o início e ocorrem em cerca de 20% dos casos.
Na recidiva, o descolamento seroso de retina do segmento posterior é menos comum do que no início, e a inflamação granulomatosa do segmento anterior predomina. Observam-se KP gorduroso, nódulos de Koeppe, nódulos de Busacca, e frequentemente ocorrem sinéquias posteriores. Sintomas prodrômicos como olho vermelho e visão turva podem aparecer.
A reação autoimune de células T CD4+ contra proteínas da melanina (família da tirosinase: tirosinase, TRP-1, TRP-2, gp100) é o núcleo da patogênese 3). Os órgãos-alvo são a úvea (coroide), sistema nervoso central (meninges), orelha interna e melanócitos da pele, sendo a inflamação granulomatosa da coroide a lesão inicial principal.
Como fator genético, é conhecida uma forte associação com HLA-DR4 (especialmente DRB10405) 3). Desequilíbrio de ligação com HLA-DPB10501 também foi relatado 3). Infecções virais (EBV, CMV) são hipotetizadas como desencadeadoras da autoimunidade por mimetismo molecular.
Principais fatores de risco:
| Tipo | Critério |
|---|---|
| Doença de VKH Completa (Complete VKH disease) | Achados oculares + achados neurológicos/auditivos + sintomas cutâneos, todos presentes |
| Doença de VKH Incompleta (Incomplete VKH disease) | Achados oculares presentes, mas ausência de achados neurológicos/auditivos ou sintomas cutâneos |
| Doença de VKH Provável (Probable VKH disease) | Apenas achados oculares (uveíte isolada) |
No início, não há manifestações cutâneas, portanto a maioria é do tipo incompleto. Posteriormente, quando surgem manifestações cutâneas, torna-se do tipo completo. O descolamento seroso bilateral da retina no início é característico, e se houver sintomas prodrômicos como cefaleia ou sintomas extraoculares como zumbido, o diagnóstico é fácil 1). Em casos atípicos (edema de papila óptica, casos unilaterais), o exame do líquido cefalorraquidiano é decisivo para o diagnóstico.
Exame do líquido cefalorraquidiano: Aumento de células com predomínio de linfócitos. Persiste por até 8 semanas. Mais útil para confirmar o diagnóstico.
Angiografia fluoresceínica (FA): Início: atraso no preenchimento coroideu com hipofluorescência em placas → Meio: vazamento puntiforme de fluoresceína → Tardio: acúmulo de corante na área de descolamento seroso da retina. Hiperfluorescência do disco óptico (cerca de 70%) 1).
Angiografia com ICG: Início: atraso no preenchimento coroideu em placas e vazamento de corante dos vasos coroideus, Meio a tardio: placas hipofluorescentes difusas. O borramento dos vasos médios e grandes devido ao distúrbio circulatório coroideu também é característico.
OCT: Detecção e monitoramento do descolamento seroso da retina. Início: espessamento coroideu acentuado. O EDI-OCT permite observar detalhadamente a secção transversal da coroide. Podem ser encontradas estruturas de membrana de fibrina e septos dentro do descolamento seroso da retina 1).
Ultrassom modo B: Espessamento coroideu. Útil para diferenciar de esclerite posterior 1).
HLA classe II: HLA-DR4 (exame auxiliar). Positividade de 80%, mas especificidade baixa (25% positivos em normais).
ERG (Eletrorretinografia): Na fase crônica, as amplitudes da visão fotópica e escotópica diminuem 3).
| Doença diferencial | Pontos de diferenciação |
|---|---|
| Esclerite posterior | Unilateral, sinal T no ultrassom, dor à movimentação ocular / dor |
| Epiteliopatia pigmentar multifocal do polo posterior (MPPE) | Unilateral, descolamento seroso vesicular migratório da retina |
| Epiteliopatia pigmentar multifocal posterior aguda (APMPPE) | Fenômeno de inversão na FA (hiperfluorescência precoce → hipofluorescência tardia), jovens, infecção prévia |
| Corioretinopatia serosa central (CSCR) | Unilateral, predominância masculina, esteroides como fator de exacerbação 3) |
| Oftalmia simpática | Diferenciado pelo histórico de trauma ocular penetrante ou cirurgia intraocular. Patologicamente considerado a mesma doença 1) |
| Síndrome de exsudação uveal idiopática | Associação com nanofthalmos, atenção ao fechamento angular |
Não é obrigatória, mas é decisiva em casos atípicos difíceis (tipo edema de papila, casos unilaterais). Como a pleocitose linfocitária persiste por até 8 semanas, a realização no momento adequado após o início aumenta a precisão diagnóstica. Em casos típicos com descolamento seroso bilateral evidente, o diagnóstico pode ser feito apenas com achados clínicos.
A pulsoterapia com esteroides seguida de redução oral gradual é o padrão. Se não tratado, muitos casos evoluem com recorrência ou persistência, levando a grave deficiência visual. Como não há indicadores na medicina atual para distinguir casos de cura espontânea dos que recorrem ou persistem, recomenda-se a administração de altas doses de esteroides sempre que possível.
Exemplo de prescrição representativa das diretrizes1):
Cronograma de redução gradual oral (comprimidos de Prednina 5 mg):
| Dose | Duração |
|---|---|
| 200 mg/dia | 2 dias |
| 150 mg/dia | 2 dias |
| 100 mg/dia | 2 dias |
| 80 mg/dia | 2 dias |
| 60 mg/dia | 4 dias |
| 40 mg/dia | 10 dias |
| 30 mg/dia | 2 semanas |
| 20 mg/dia | 4 semanas |
| 15 mg/dia | 4 semanas |
| 10 mg/dia | 4 semanas |
| 5 mg/dia | 4 semanas |
| 5 mg/dia (em dias alternados) | 4 semanas |
A redução é feita lentamente, e a medicação é suspensa ao longo de 6 meses ou mais, mesmo sem recidiva. Com tratamento inferior a 6 meses, a taxa de recidiva é de 58,8%; com 6 meses ou mais, cai para 11,1%5). Em caso de recidiva, o corticosteroide é aumentado e reduzido gradualmente por um período mais longo do que da vez anterior. Com a monoterapia com corticosteroide, 44% apresentam recidiva e 59% desenvolvem fundo em “olho de sol poente” (sunset glow fundus)2).
Dose para pacientes pediátricos: Em pacientes com idade e peso adequados, administrar metilprednisolona 500 mg/dia por via intravenosa por 3 dias (terapia de pulso) ou prednisolona 0,5–1,0 mg/kg/dia por via oral com redução gradual. A redução é feita com atenção aos efeitos colaterais.
Ciclosporina (Neoral®): 3 mg/kg/dia (para peso de 60 kg: 180 mg/dia dividido em 2 doses). Necessário monitoramento regular da concentração basal. Espera-se redução da dose de corticosteroide. Aplicado em casos persistentes1). Atenção a efeitos colaterais como suscetibilidade a infecções, disfunção renal e hepática.
Metotrexato (MTX): 25 mg/semana por via oral. Iniciar com 15 mg/semana por 2 semanas e depois aumentar2).
Micofenolato de mofetila (MMF): 1,5 g duas vezes ao dia por via oral. Iniciar com 500 mg duas vezes ao dia e aumentar gradualmente2).
O estudo FAST (NCT01829295) é um ECR para uveíte não infecciosa (subanálise de 93 casos de VKH de 216 casos). 49 casos foram randomizados para MTX e 44 para MMF, iniciando com prednisona 1 mg/kg/dia (máx 60 mg/dia) com redução gradual (alvo ≤7,5 mg/dia aos 6 meses).
Desfecho Primário de 6 Meses (Subanálise de VKH):
| Indicador | Grupo MTX | Grupo MMF | Valor P |
|---|---|---|---|
| Taxa de Sucesso do Tratamento | 80,4% (37/46 casos) | 64,1% (25/39 casos) | 0,10 |
| Redução da Espessura Retiniana Foveal | −62,5 μm | −4,0 μm | 0,003 |
| Taxa de Desaparecimento de SRD | 86,3% | 64,1% | 0,02 |
| Melhora da visão | Equivalente | Equivalente | 0.78 |
A taxa geral de sucesso do tratamento é de 74,7% (62/85 casos). Na VKH aguda, MTX foi superior na redução da CST e desaparecimento do SRD. Aos 12 meses, 91,3% do grupo MMF mantiveram o sucesso do tratamento, e cerca de metade (MTX 50,0%, MMF 56,5%) alcançaram a suspensão completa da prednisona 2). Na falha do tratamento inicial e mudança de MMF para MTX, 81,8% tiveram sucesso 2). Com o uso precoce de MMF, 93% dos casos agudos de VKH mantiveram 20/20, e não houve relato de recidiva ou formação de sunset glow fundus 2).
Em casos difíceis de administração sistêmica de esteroides (idosos, gestantes, diabéticos, histórico de doença mental), considerar injeção sub-Tenon posterior de triancinolona acetonida 1).
Catarata comórbida: A catarata esteroide aparece com alta frequência devido ao uso intenso de esteroides. Casos de remissão completa têm o mesmo risco da cirurgia habitual. A implantação de LIO também não é problema. Se houver possibilidade de cirurgia filtrante futura, optar por preservação conjuntival superior e incisão corneana.
Glaucoma secundário e glaucoma esteroide: Usar colírios hipotensores (betabloqueadores, análogos de PG, inibidores da anidrase carbônica) → inibidores da anidrase carbônica orais → infusão de D-manitol, nessa ordem. Realizar trabeculotomia (eficaz no glaucoma esteroide), se insuficiente, realizar trabeculectomia.
Recomenda-se a redução gradual lenta por 6 meses ou mais, mesmo sem recidiva. A interrupção antes de 6 meses resulta em alta taxa de recidiva de cerca de 58,8%, enquanto a continuação por mais de 6 meses reduz para 11,1% 5). Em caso de recidiva, reduzir mais lentamente do que da vez anterior e considerar a adição de imunossupressores.
A ciclosporina (Neoral® 3 mg/kg/dia) é usada como poupadora de esteroides em casos persistentes 1). O estudo FAST confirmou a eficácia do metotrexato (MTX 25 mg/semana) e do micofenolato de mofetila (MMF 1,5 g duas vezes/dia). Na VKH aguda, o MTX mostrou tendência superior na redução da espessura retiniana foveal e na taxa de desaparecimento do SRD 2).
A reação autoimune de células T CD4+ contra proteínas da melanina (tirosinase, TRP-1, TRP-2, gp100) é a patogênese central 3). Os alvos são melanócitos na úvea (coroide), sistema nervoso central (meninges), orelha interna e pele; a inflamação granulomatosa da coroide é a lesão inicial principal.
Fatores genéticos mostram forte associação com HLA-DR4 (especialmente DRB10405) 3), e também foi relatado desequilíbrio de ligação com HLA-DPB10501 3). Cerca de metade dos casos apresenta sintomas gripais nas duas semanas antes do início, sugerindo que infecção viral (EBV, CMV) desencadeia autoimunidade por mimetismo molecular.
Interpretação de Imagens: Na ICG, observam-se borramento de vasos médios-grandes e manchas hipofluorescentes devido a distúrbio circulatório coroidal. O espessamento coroidal acentuado precoce na EDI-OCT reflete edema do estroma coroidal. A perda crônica de melanina forma o fundo em “pôr do sol” 3).
Mesmo com pulsoterapia com esteroides, a inflamação persiste em cerca de 25% dos casos, levando a atrofia retinocoroidal progressiva e perda visual grave em alguns casos. Frequentemente permanecem sintomas subjetivos como leve distorção ou anormalidade de visão de cores.
Significância Clínica do Estudo FAST 2): Primeiro ECR comparando diretamente MTX e MMF em uveíte não infecciosa. 93 casos de VKH foram o maior subgrupo do estudo, mostrando eficácia da introdução precoce de imunossupressor na VKH aguda. Estudos de acompanhamento são esperados.
Importância da Introdução Precoce de Imunossupressor 2): Com esteroide isolado, 44% recidivam e 59% desenvolvem fundo em “pôr do sol”. A combinação precoce de esteroide + antimetabólito pode reduzir as taxas de recidiva e de fundo em “pôr do sol”. O conceito de “janela terapêutica” — iniciar esteroide precocemente na fase aguda para prevenir progressão para fase crônica recidivante e reduzir necessidade de imunossupressão prolongada — está se difundindo.
Evolução do Diagnóstico por Imagem3): A avaliação quantitativa da espessura coroidal por EDI-OCT e a detecção de coroidite latente por ICG são utilizadas no monitoramento do tratamento. A medição quantitativa dos focos coroidais hiper-refletivos (HCF) por en face OCT pode ser um biomarcador para avaliar a atividade da VKH.
Tendências de Revisão dos Critérios Diagnósticos2): Foi proposta uma classificação precoce/tardia incorporando OCT, FA e ICG, e espera-se melhora na precisão diagnóstica. Há observações de que os critérios revisados de 2001 têm limitações na detecção precoce da VKH.