Etapa prodrómica
Momento: 1–2 semanas antes de que aparezcan los síntomas oculares
Síntomas: Hormigueo en el cuero cabelludo, dolor de cabeza (64.7%), tinnitus (24.7%), malestar general (21.1%), síntomas similares a los de la gripe2)

La enfermedad de Vogt-Koyanagi-Harada (enfermedad de VKH) es una enfermedad autoinmune mediada por células T contra proteínas de melanina. En la fase de inicio, se presenta con uveítis, síntomas de irritación meníngea como cefalea y pérdida auditiva. A medida que la inflamación persiste, aparecen manifestaciones cutáneas como vitíligo, alopecia y poliosis. Las lesiones inflamatorias ocurren en todo el cuerpo donde se distribuyen los melanocitos: ojos, cabello, piel, oído interno y meninges.
Aproximadamente la mitad de los pacientes presentan síntomas similares a los de la gripe dentro de las 2 semanas previas al inicio, lo que sugiere que una infección viral puede desencadenar la enfermedad a través de reactividad cruzada inmunológica (mimetismo molecular). Se ha sugerido la participación del virus de Epstein-Barr y el citomegalovirus. El pronóstico visual es generalmente bueno, pero algunos casos progresan a deterioro visual severo debido a la inflamación persistente, por lo que el diagnóstico temprano y el tratamiento inicial son importantes.
La VKH es la segunda causa más común de uveítis. En una encuesta epidemiológica de 2002 se reportaron 205 casos de VKH (6.7%), y en una encuesta de 2009, 267 casos (7.0%), ambos ocupando el segundo lugar después de la sarcoidosis1). La edad típica de inicio es entre los 20 y 50 años, con un ligero predominio femenino. La positividad para HLA-DR4 se encuentra en el 80% de los pacientes (en comparación con el 25% en la población japonesa normal), y es más común en personas de color (asiáticos, hispanos, nativos americanos) y rara en caucásicos. Entre 93 pacientes con VKH inscritos en el ensayo FAST, el 71% eran mujeres, con una mediana de edad de 35 a 38 años2). Es más común en el este de Asia, pero también ocurre en las regiones del Pacífico de América del Norte y del Sur3).
Es más común en personas de color (asiáticos, hispanos, nativos americanos) y rara en caucásicos. Tiende a ocurrir en individuos HLA-DR4 positivos y en aquellos de 20 a 50 años, con un ligero predominio femenino. Se cree que se desarrolla a partir de una combinación de predisposición genética (HLA-DRB1*0405) y algún desencadenante ambiental3).
Etapa prodrómica
Momento: 1–2 semanas antes de que aparezcan los síntomas oculares
Síntomas: Hormigueo en el cuero cabelludo, dolor de cabeza (64.7%), tinnitus (24.7%), malestar general (21.1%), síntomas similares a los de la gripe2)
Etapa aguda (inicio ocular)
Pérdida aguda de la visión: Desprendimiento seroso de retina bilateral (57.6% de VKH agudo)2)
Inflamación leve de la cámara anterior y opacidad vítrea, hiperemia e hinchazón del disco óptico (aproximadamente 70%)
Pliegues coroideos: En casos graves, desprendimiento coroideo y cámara anterior poco profunda
Etapa crónica (etapa recurrente)
Predomina la inflamación granulomatosa del segmento anterior
KP en grasa de cerdo, nódulos de Koeppe/Busacca, sinequias posteriores
El fondo de ojo en rojo atardecer aparece en el 33.3% de los VKH crónicos2)
Etapa tardía (convalecencia)
Fondo de ojo en rojo atardecer (sunset glow fundus): pérdida de melanina coroidea
Signo de Sugiura: despigmentación del limbo (aproximadamente 1 mes después del inicio)
Manifestaciones cutáneas (aproximadamente 20%): vitíligo, alopecia, canicie (después de varios meses)
Acumulación de pigmento macular, manchas despigmentadas coriorretinianas dispersas
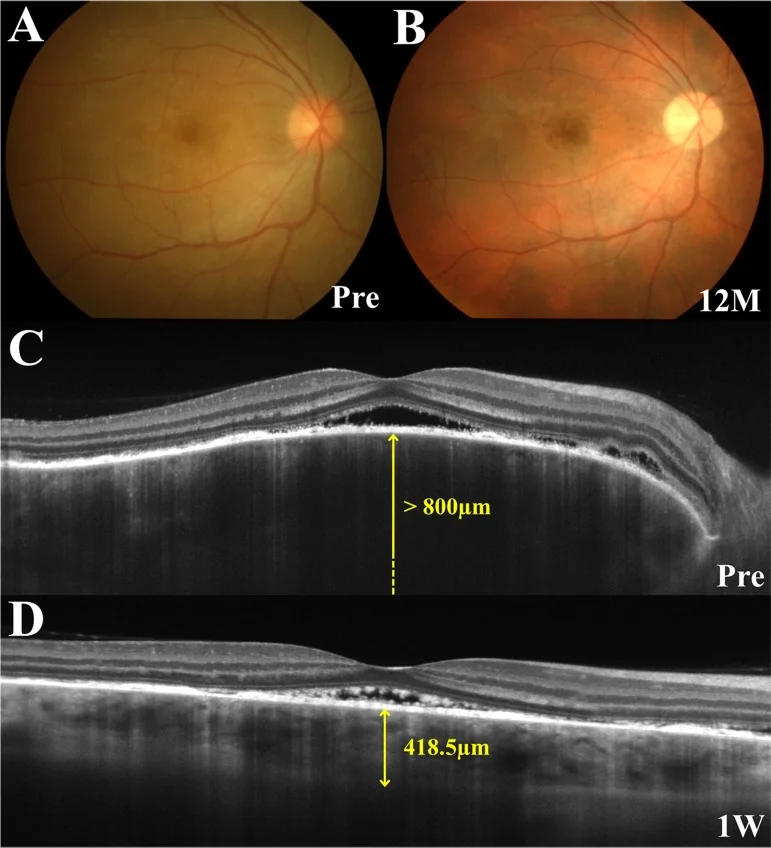
En la etapa prodrómica aparecen parestesia del cuero cabelludo, cefalea y tinnitus, seguidos 1 a 2 semanas después de una disminución aguda de la visión y visión borrosa en ambos ojos simultánea o secuencialmente. En la OCT, es característico observar estructuras septales dentro del desprendimiento de retina en la fase aguda.
La irritación meníngea (cefalea, rigidez de nuca) aparece en la mayoría de los casos al inicio de la enfermedad. Hipoacusia neurosensorial (pocos síntomas subjetivos pero detectable en pruebas). Las manifestaciones cutáneas (vitíligo, alopecia, canicie) aparecen varios meses después del inicio y se observan en aproximadamente el 20% de los pacientes.
En la recurrencia, el desprendimiento seroso de retina del segmento posterior, como en el episodio inicial, es menos frecuente, y predomina la inflamación granulomatosa del segmento anterior. Se observan precipitados queráticos en grasa de cerdo (KP), nódulos de Koeppe y nódulos de Busacca, y a menudo se producen sinequias posteriores. Los síntomas prodrómicos pueden incluir enrojecimiento ocular y visión borrosa.
El núcleo de la patología es una reacción autoinmune de células T CD4-positivas contra las proteínas de melanina (familia de la tirosinasa: tirosinasa, TRP-1, TRP-2, gp100) 3). Los órganos diana son los melanocitos de la úvea (coroides), el sistema nervioso central (meninges), el oído interno y la piel, siendo la inflamación granulomatosa de la coroides la lesión principal inicial.
Se conoce una fuerte asociación con HLA-DR4 (especialmente DRB10405) como predisposición genética 3). También se ha reportado desequilibrio de ligamiento con HLA-DPB10501 3). Existe la hipótesis de que las infecciones virales (VEB, CMV) desencadenan la autoinmunidad mediante mimetismo molecular.
Principales factores de riesgo:
| Tipo | Criterios |
|---|---|
| Enfermedad de VKH completa | Hallazgos oculares + hallazgos neurológicos/auditivos + hallazgos cutáneos, todos presentes |
| Enfermedad de VKH incompleta | Hallazgos oculares presentes, falta hallazgos neurológicos/auditivos o cutáneos |
| Enfermedad de VKH probable | Solo hallazgos oculares (uveítis aislada) |
En la etapa temprana del inicio, no se observan hallazgos cutáneos, por lo que la mayoría son del tipo incompleto. Más tarde, cuando aparecen los hallazgos cutáneos, se convierte en el tipo completo. El desprendimiento seroso de retina bilateral en la etapa temprana es característico, y el diagnóstico es fácil si hay síntomas prodrómicos como cefalea o síntomas extraoculares como tinnitus 1). En casos atípicos (tipo edema de papila óptica, casos unilaterales), el examen del líquido cefalorraquídeo es decisivo para el diagnóstico.
Examen del líquido cefalorraquídeo: Pleocitosis de predominio linfocítico. Persiste hasta 8 semanas. Más útil para confirmar el diagnóstico.
Angiografía fluoresceínica (FA): En la fase temprana muestra hipofluorescencia parcheada por retraso en el llenado coroideo → fase media muestra hiperfluorescencia granular (fugas puntiformes) → fase tardía muestra acumulación de colorante en el área del desprendimiento seroso de retina. Hiperfluorescencia del disco óptico (aproximadamente 70%) 1).
Angiografía con ICG: En la fase temprana muestra retraso en el llenado coroideo parcheado y fuga de colorante de los vasos coroideos; en las fases media y tardía muestra manchas hipofluorescentes dispersas. El desenfoque de los vasos medianos y grandes debido a la alteración de la circulación coroidea también es característico.
OCT: Detección y monitorización del desprendimiento seroso de retina. Engrosamiento coroideo marcado en la etapa temprana. La EDI-OCT permite observar en detalle el corte transversal de la coroides. Pueden observarse estructuras membranosas similares a fibrina o tabiques dentro del desprendimiento seroso de retina 1).
Ecografía modo B: Hallazgos de engrosamiento coroideo. Útil para diferenciar de escleritis posterior 1).
HLA clase II: HLA-DR4 (prueba auxiliar). Tasa de positividad del 80%, pero baja especificidad (25% positivo en individuos normales).
ERG (electrorretinograma): En la fase crónica, las amplitudes de las respuestas fotópicas y escotópicas disminuyen 3).
| Enfermedad Diferencial | Puntos Diferenciales |
|---|---|
| Escleritis posterior | Unilateral, signo de T en ecografía, dolor con los movimientos oculares/dolor |
| Epiteliopatía pigmentaria posterior múltiple (MPPE) | Unilateral, desprendimiento seroso de retina ampolloso migratorio |
| Epiteliopatía pigmentaria placoidal multifocal posterior aguda (APMPPE) | Fenómeno de inversión en la angiografía fluoresceínica (hiperfluorescencia temprana → hipofluorescencia tardía), edad joven, infección previa |
| Coriorretinopatía serosa central (CSCR) | Unilateral, predominio masculino, los esteroides como factor agravante3) |
| Oftalmía simpática | Se diferencia por antecedentes de traumatismo ocular penetrante o cirugía intraocular. Patológicamente se considera la misma enfermedad1) |
| Síndrome de efusión uveal idiopática | Asociación con nanoftalmos, precaución por cierre angular |
No es obligatorio, pero puede ser decisivo en casos atípicos de difícil diagnóstico (tipo edema de papila óptica, casos unilaterales). Dado que la pleocitosis linfocítica persiste hasta 8 semanas, realizarlo en el momento adecuado después del inicio mejora la precisión diagnóstica. En casos típicos con desprendimiento seroso de retina bilateral evidente, a menudo se puede diagnosticar solo con los hallazgos clínicos.
La terapia de pulso de esteroides seguida de reducción gradual oral en dosis altas es el estándar. Si no se trata y se observa, muchos casos presentan recurrencia o persistencia que conducen a una discapacidad visual grave. Dado que la medicina actual no tiene un indicador para distinguir los casos que se curan espontáneamente de los que recurren o persisten, se recomienda la administración de dosis altas de esteroides cuando sea posible.
Ejemplo de prescripción de caso representativo de la guía1):
Programa de reducción oral (comprimidos de Predonina 5 mg):
| Dosis | Duración |
|---|---|
| 200 mg/día | 2 días |
| 150 mg/día | 2 días |
| 100 mg/día | 2 días |
| 80 mg/día | 2 días |
| 60 mg/día | 4 días |
| 40 mg/día | 10 días |
| 30 mg/día | 2 semanas |
| 20 mg/día | 4 semanas |
| 15 mg/día | 4 semanas |
| 10 mg/día | 4 semanas |
| 5 mg/día | 4 semanas |
| 5 mg/día (días alternos) | 4 semanas |
La reducción debe hacerse lentamente, y aunque no haya recurrencia, la suspensión debe tomar al menos 6 meses. El tratamiento de menos de 6 meses tiene una tasa de recurrencia del 58.8%, mientras que el de 6 meses o más la reduce al 11.1% 5). Si hay recurrencia, aumente la dosis de esteroides y reduzca más lentamente que antes. Con la monoterapia con esteroides, el 44% recae y el 59% desarrolla fondo de ojo en rojo atardecer 2).
Dosis de tratamiento en pacientes pediátricos: En niños de cierta edad y peso, se administra metilprednisolona 500 mg/día IV durante 3 días (terapia de pulso) o prednisolona oral comenzando con 0.5–1.0 mg/kg/día con reducción gradual. Reduzca mientras vigila los efectos secundarios.
Ciclosporina (Neoral®): 3 mg/kg/día (para 60 kg de peso: 180 mg/día en dos dosis divididas). Se requiere monitorización regular de la concentración valle. Se espera que reduzca la dosis de esteroides. Se usa en casos refractarios 1). Vigile los efectos secundarios como mayor susceptibilidad a infecciones, disfunción renal y disfunción hepática.
Metotrexato (MTX): 25 mg/semana por vía oral. Comience con 15 mg/semana durante 2 semanas, luego aumente 2).
Micofenolato de mofetilo (MMF): 1.5 g dos veces al día por vía oral. Comience con 500 mg dos veces al día y aumente gradualmente 2).
El ensayo FAST (NCT01829295) es un ECA para uveítis no infecciosa (subanálisis de 93 casos de VKH de 216). 49 pacientes fueron asignados al azar a MTX y 44 a MMF, comenzando con prednisona 1 mg/kg/día (máx. 60 mg/día) y reduciendo (objetivo ≤7.5 mg/día a los 6 meses).
Resultado principal a los 6 meses (subanálisis de VKH):
| Indicador | Grupo MTX | Grupo MMF | Valor P |
|---|---|---|---|
| Tasa de éxito del tratamiento | 80.4% (37/46) | 64.1% (25/39) | 0.10 |
| Reducción del grosor retiniano foveal | −62.5 μm | −4.0 μm | 0.003 |
| Tasa de resolución de SRD | 86.3% | 64.1% | 0.02 |
| Mejoría visual | Equivalente | Equivalente | 0.78 |
La tasa de éxito del tratamiento global fue del 74.7% (62/85 casos). En VKH agudo, MTX fue superior en la reducción de CST y la desaparición de SRD. A los 12 meses, el 91.3% del grupo MMF continuó con éxito el tratamiento, y aproximadamente la mitad (MTX 50.0%, MMF 56.5%) logró la suspensión completa de prednisona 2). El cambio de MMF a MTX tras el fracaso del tratamiento inicial tuvo éxito en el 81.8% de los casos 2). Con la combinación inicial de MMF, el 93% de los pacientes con VKH agudo mantuvo una visión de 20/20, y todos los casos no presentaron recurrencia ni formación de fondo de ojo en atardecer 2).
En casos difíciles para la administración sistémica de esteroides (ancianos, embarazadas, diabetes, antecedentes de enfermedad psiquiátrica), considerar la inyección subtenoniana posterior de triamcinolona acetonida 1).
Catarata complicada: El uso intensivo de esteroides produce una alta tasa de catarata esteroidea. En casos de remisión completa, el riesgo es similar al de la cirugía normal. La implantación de LIO también es segura. Si es posible que se necesite una cirugía de filtración más adelante, elija la preservación conjuntival superior y la incisión corneal.
Glaucoma secundario / glaucoma esteroideo: Use gotas antihipertensivas (betabloqueantes, análogos de prostaglandinas, inhibidores de la anhidrasa carbónica) → CAI oral → infusión intravenosa de D-manitol en ese orden. Realice una trabeculotomía (eficaz para el glaucoma esteroideo) y, si es insuficiente, una trabeculectomía.
Incluso sin recurrencia, se recomienda reducir gradualmente durante al menos 6 meses. La suspensión en menos de 6 meses da una tasa de recurrencia alta de aproximadamente 58.8%, mientras que la continuación durante 6 meses o más la reduce al 11.1% 5). En caso de recurrencia, reduzca más lentamente que la vez anterior y considere agregar fármacos inmunosupresores.
La ciclosporina (Neoral® 3 mg/kg/día) se utiliza como fármaco ahorrador de esteroides en casos refractarios 1). El ensayo FAST confirmó la eficacia del metotrexato (MTX 25 mg/semana) y el micofenolato de mofetilo (MMF 1,5 g dos veces al día). En la VKH aguda, el MTX mostró una tendencia a una mayor reducción del grosor retiniano foveal y de la tasa de desaparición del SRD 2).
La patología central es una respuesta autoinmune de células T CD4 positivas contra las proteínas de melanina (tirosinasa, TRP-1, TRP-2, gp100) 3). Los objetivos son los melanocitos de la úvea (coroides), el sistema nervioso central (meninges), el oído interno y la piel. La inflamación granulomatosa de la coroides es la lesión principal inicial.
Se conoce una fuerte asociación genética con HLA-DR4 (especialmente DRB10405) 3), y también se ha informado desequilibrio de ligamiento con HLA-DPB10501 3). Aproximadamente la mitad de los pacientes presentan síntomas similares a los de un resfriado dentro de las dos semanas previas al inicio, lo que sugiere que las infecciones virales (VEB, CMV) pueden desencadenar autoinmunidad mediante mimetismo molecular.
Interpretación de hallazgos de imagen: La ICG muestra borrosidad de los vasos medianos y grandes y manchas hipofluorescentes debido a la alteración circulatoria coroidea. El engrosamiento coroideo temprano marcado en EDI-OCT refleja edema del estroma coroideo. En la fase crónica, la pérdida de melanina conduce a un fondo de ojo en atardecer 3).
Incluso con la terapia de pulsos de esteroides, aproximadamente el 25% de los casos presentan inflamación persistente, y la atrofia coriorretiniana gradual puede provocar un deterioro visual severo en algunos pacientes. A menudo persisten síntomas subjetivos leves como metamorfopsia y anomalías de la visión cromática.
Importancia clínica del ensayo FAST 2): Este es el primer ECA de comparación directa de MTX vs. MMF en uveítis no infecciosa. Los 93 casos de VKH fueron el subgrupo más grande, demostrando la eficacia de la introducción temprana de inmunosupresores en la VKH aguda. Se esperan más estudios de seguimiento.
Importancia de la introducción temprana de inmunosupresores 2): Con la monoterapia con esteroides, el 44% recae y el 59% desarrolla fondo de ojo en atardecer. La combinación temprana de esteroides con antimetabolitos puede reducir las tasas de recaída y la incidencia de fondo de ojo en atardecer. El concepto de “ventana terapéutica” —iniciar esteroides temprano en la enfermedad para prevenir la progresión a la fase crónica recurrente y reducir la necesidad de inmunosupresión a largo plazo— está ganando aceptación.
Evolución del diagnóstico por imagen3): La evaluación cuantitativa del engrosamiento coroideo mediante EDI-OCT y la detección de coroiditis oculta mediante ICG se utilizan para el monitoreo del tratamiento. La medición cuantitativa de los focos coroideos hiperreflectivos (HCF) mediante OCT en face podría convertirse en un biomarcador para la evaluación de la actividad de VKH.
Tendencias en los criterios diagnósticos revisados2): Se ha propuesto una clasificación en etapas temprana/tardía que incorpora OCT, FA e ICG, y se espera una mejora en la precisión diagnóstica. Se ha señalado que los criterios revisados de 2001 actuales tienen limitaciones para detectar VKH temprana.