L’epiteliopatia pigmentaria placoida multifocale posteriore acuta (acute posterior multifocal placoid pigment epitheliopathy; APMPPE) è una malattia infiammatoria acuta bilaterale descritta per la prima volta da Gass nel 1968 10). È caratterizzata da multiple macchie bianche discoidi a livello degli strati profondi della retina e dell’epitelio pigmentato retinico (RPE) al polo posteriore di entrambi gli occhi.
Secondo la classificazione delle linee guida cliniche per l’uveite (Jpn J Ophthalmol 2019;123(6):635-696), è classificata come uveite posteriore non infettiva a predominanza di lesioni del fondo oculare, con caratteristiche cliniche di bilateralità, esordio acuto e decorso transitorio 1).
Il meccanismo patogenetico ipotizzato è una vasculite obliterante dovuta a una reazione di ipersensibilità ritardata (allergia di tipo IV) nelle arteriole afferenti della coriocapillare. Si sospetta un’infezione virale (influenza, parotite, ecc.) come fattore scatenante, ma i dettagli sono sconosciuti.
L’età di predilezione è 20-30 anni (media 25 anni), senza differenze di sesso 1). Spesso bilaterale. In più della metà dei casi vengono riportati sintomi simil-influenzali precedenti 1). Raramente è stata riportata un’associazione con vasculite sistemica, nefropatia, meningoencefalite, tiroidite 1).
QL'APMPPE si verifica solo nei giovani?
A
L’età di predilezione sono i giovani adulti sotto i 40 anni; l’insorgenza negli anziani o nei bambini è rara. Se lesioni simili si osservano dopo i 40 anni, è importante la differenziazione dalla coroidite serpiginosa (coroidite geografica).
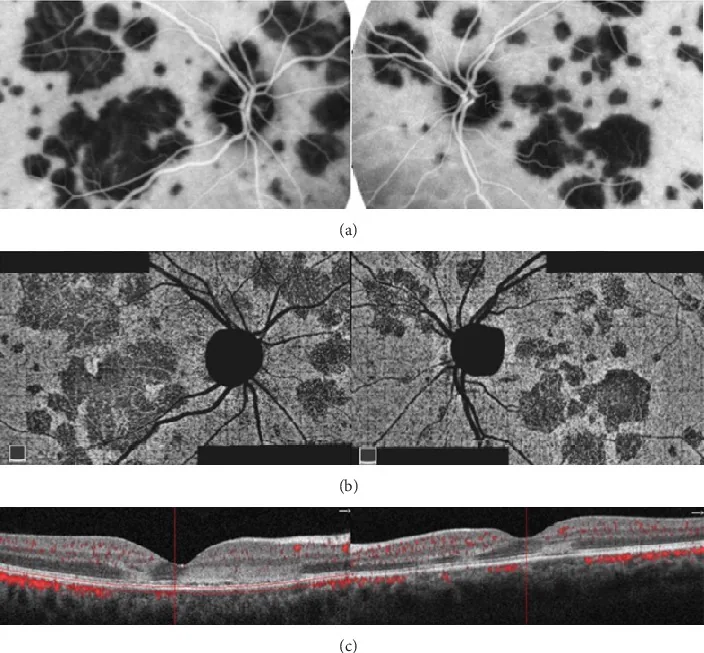
Oliveira MA, et al. Management of Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE): Insights from Multimodal Imaging with OCTA. Case Rep Ophthalmol Med. 2020. Figure 5. PMCID: PMC7094199. License: CC BY.
Al polo posteriore di entrambi gli occhi si osservano lesioni geografiche e maculari multiple, che appaiono ipofluorescenti all’ICGA e come aree di ridotto flusso sanguigno nella coriocapillare corrispondente all’OCTA. L’OCT mostra alterazioni iperriflettenti dallo strato plessiforme esterno all’EPR, rappresentando i reperti tipici dell’APMPPE.
Al polo posteriore di entrambi gli occhi compaiono multiple macchie bianche disciformi grigio-biancastre (color crema-bianco) negli strati profondi della retina fino al livello dell’EPR. Le lesioni sono ben delimitate, di dimensioni quasi uniformi (1/4–1/2 diametro papillare), da poche a numerose, con scarsa tendenza a confluire o espandersi. Si possono osservare lieve arrossamento ed edema della papilla ottica e lievi cellule vitreali 1). Un punto di differenziazione è che sono più grandi delle macchie bianche della MEWDS.
Reperti in fase acuta
Macchie bianche del fondo: Multiple macchie bianche disciformi grigio-biancastre ben delimitate di 1/4–1/2 diametro papillare al polo posteriore. Edema ischemico e opacità degli strati retinici esterni e dell’EPR dovuti all’occlusione della coriocapillare.
Danno degli strati retinici esterni: L’OCT mostra un’alterazione iperriflettente della zona ellissoidale (linea IS/OS) 1). La distruzione della zona ellissoidale causa sintomi soggettivi 2).
FAF (autofluorescenza del fondo): In fase acuta, le lesioni appaiono ipo- o iperautofluorescenti. I reperti dell’OCT e dell’autofluorescenza sono strettamente correlati 9).
Reperti in fase di remissione
Regressione delle macchie bianche: Le macchie bianche regrediscono dal centro in 7–12 giorni e scompaiono lasciando lievi alterazioni di depigmentazione.
Recupero OCT: La zona ellissoidale può recuperare in alcuni mesi, ma può persistere un assottigliamento dello strato dei fotorecettori 1).
Atrofia corioretinica: in alcuni casi persistono lievi chiazze di atrofia corioretinica1).
QLe macchie bianche dell'APMPPE scompaiono?
A
Nella maggior parte dei casi, le macchie bianche regrediscono entro 7-12 giorni dall’esordio e scompaiono lasciando una depigmentazione. La prognosi visiva è generalmente buona e le recidive sono rare. Tuttavia, nei casi di transizione verso la corioretinopatia geografica (coroidite serpiginosa), si osserva una progressione, pertanto è necessario un follow-up regolare.
Si ipotizza che una vasculite occlusiva dovuta a una reazione di ipersensibilità ritardata (allergia di tipo IV) delle arteriole afferenti della coriocapillare sia alla base della malattia. L’OCT-A (angiografia OCT) mostra un flow void (assenza di segnale di flusso) a livello della coriocapillare nelle lesioni acute, supportando l’ischemia della coriocapillare come nucleo della patologia3). Klufas et al. hanno riportato che l’OCT-A rileva il flow void della coriocapillare con un’elevata concordanza con FA e ICGA in tre malattie della corioretinite a placca (APMPPE, PPM, RPC)4).
Esistono casi che si verificano dopo sintomi simil-influenzali (influenza, parotite, ecc.), infezione delle vie respiratorie superiori o vaccinazione.
Sono state riportate associazioni con vasculite sistemica (vasculite cerebrale), nefropatia, poliarterite nodosa e tiroidite1).
QL'APMPPE comporta un rischio di ictus?
A
Sebbene rari, sono stati riportati casi di APMPPE complicata da vasculite del sistema nervoso centrale. In caso di comparsa di sintomi neurologici (forte mal di testa, alterazione della coscienza, paralisi), è necessario eseguire d’urgenza una RM e un’ARM cerebrale e collaborare con un neurologo.
La diagnosi di APMPPE si basa sui reperti caratteristici del fondo oculare e sui risultati dell’angiografia con fluoresceina (FA, ICGA). L’imaging multimodale consente una valutazione multidimensionale della patologia 2).
Il ‘fenomeno di inversione della fluorescenza’ (ipotofluorescenza precoce → iperfluorescenza tardiva) osservato all’angiografia con fluoresceina (FA) è il reperto più caratteristico dell’APMPPE. Viene interpretato come un blocco precoce del flusso di colorante nella coriocapillare (a causa di una vasculite occlusiva) e una successiva iperfluorescenza tardiva per essudazione dalle aree circostanti 1). All’angiografia con verde indocianina (ICGA) si osserva ipotofluorescenza persistente dalla fase precoce a quella tardiva, che riflette più direttamente l’ischemia della coriocapillare5).
L’OCT-A è una modalità di imaging non invasiva per valutare i disturbi del flusso sanguigno nella coriocapillare nell’APMPPE. Furino et al. hanno riportato che l’OCT-A è in grado di rilevare i flow void della coriocapillare con un alto tasso di concordanza con FA e ICGA nelle lesioni acute di APMPPE 8). Nella fase di recupero si osserva una riduzione dei flow void insieme a un miglioramento della funzione visiva, rendendolo un biomarcatore utile per il monitoraggio dell’attività.
Maculopatia placoida persistente (PPM) / corioretinite placoida relent (RPC): Forma simile all’APMPPE, oggetto di discussione per una riclassificazione nello ‘spettro delle corioretiniti placoidi’ 4)7)
QCome distinguere l'APMPPE dal MEWDS?
A
Tre punti chiave sono la dimensione delle lesioni, i reperti alla FA e la bilateralità. Le macchie bianche dell’APMPPE sono più grandi di quelle del MEWDS (1/4–1/2 diametro papillare), spesso bilaterali, e mostrano alla FA un fenomeno di inversione (ipotofluorescenza precoce seguita da iperfluorescenza tardiva). Il MEWDS è spesso unilaterale, con macchie più piccole e diffuse, e mostra iperfluorescenza precoce alla FA1).
La tendenza alla risoluzione spontanea è forte e la guarigione avviene spesso senza trattamento specifico 1). Nei casi lievi, l’osservazione è la regola. Anche il rapporto di follow-up a lungo termine di Jones (1995) mostra che molti casi hanno infine recuperato una buona acuità visiva10).
In caso di significativa riduzione dell’acuità visiva, complicanza da papillite o lesioni vicino alla macula, considerare quanto segue.
Compresse di prednisolone (5 mg): iniziare con 30 mg/die, poi ridurre gradualmente nell’arco di 2 settimane-1 mese
Compresse di Carnaculina (callidinogenasi 25-50 unità): 3 compresse in 3 dosi (come coadiuvante per migliorare la microcircolazione coroidale)
Nei casi di transizione verso una coroidopatia geografica o complicanza da vasculite del SNC, considerare la terapia pulsata con steroidi (metilprednisolone ad alte dosi per via endovenosa) o l’uso di immunosoppressori 1). In caso di neovascolarizzazione coroidale (CNV), considerare l’iniezione intravitreale di anti-VEGF, ma si tratta di una complicanza rara. Nei casi di transizione verso una maculopatia placoida persistente (PPM), può essere necessaria una gestione a lungo termine, come sottolineato da Kolomeyer e Brucker nella loro revisione sistematica 7).
QIl trattamento steroideo è sempre necessario?
A
Nei casi lievi, la sola osservazione è sufficiente per la guarigione spontanea. La terapia steroidea viene presa in considerazione in caso di significativa riduzione dell’acuità visiva, grave infiammazione vicino alla papilla o sintomi del SNC. Anche le linee guida per la gestione delle uveiti indicano che la “guarigione spontanea senza trattamento specifico” è il principio di base 1).
La patologia dell’APMPPE è intesa come un cambiamento ischemico centrato sull’occlusione della coriocapillare.
Si ipotizza che una vasculite obliterante dovuta a una reazione di ipersensibilità ritardata (allergia di tipo IV) nelle arteriole afferenti della coriocapillare sia la causa sottostante. Occlusione → alterazioni ischemiche dell’EPR e degli strati esterni della retina (edema, opacità) → formazione di macchie bianche discoidi. Mrejen et al. hanno mostrato che le lesioni coroidali si estendono più in profondità, suggerendo un possibile coinvolgimento non solo della coriocapillare ma anche dei vasi medi e grandi 3).
Nell’OCT-A si osservano aree di flow void a livello della coroide interna, ed è stato dimostrato che l’ischemia della coriocapillare è costantemente presente come base patofisiologica del gruppo di malattie dello ‘spettro dei disordini placoidi’ (inclusi la maculopatia placoide persistente e la corioretinite placoide implacabile) simile all’APMPPE 4). Klufas et al. hanno riportato che le tre malattie mostrano un pattern comune di flow void della coriocapillare all’OCT-A, supportando il concetto di spettro della corioretinite placoide.
Meccanismo del fenomeno di inversione della fluorescenza: nella fase precoce del contrasto, l’afflusso di colorante nella coriocapillare è ostacolato, causando ipofluorescenza; nella fase tardiva, il colorante fuoriesce dai tessuti normali circostanti, trasformandosi in iperfluorescenza. Il fatto che l’ICGA mostri ipofluorescenza per tutto il tempo riflette anche i cambiamenti occlusivi della coriocapillare5). La FAF consente di valutare in modo non invasivo la distribuzione del danno all’EPR e mostra un’elevata correlazione con i reperti OCT9).
APMPPE dopo infezione/vaccinazione COVID-19: Sono stati riportati casi di lesioni acute simil-APMPPE dopo l’infezione, e si sta prestando attenzione alla relazione con la risposta immunitaria indotta dal virus.
Complicanza di vasculite cerebrale: La raccomandazione per la risonanza magnetica/angiografia cerebrale nei pazienti con sintomi neurologici si sta rafforzando. Sono stati riportati diversi casi di APMPPE complicata da ictus o vasculite cerebrale, sottolineando l’importanza del monitoraggio sistemico oltre ai sintomi oculari 6).
Progressi nell’imaging multimodale: Il monitoraggio del flow void della coriocapillare mediante OCT-A potrebbe diventare un biomarcatore per la valutazione dell’attività 8). Essendo non invasiva e ripetibile, sta diventando una modalità importante che integra l’angiografia con fluoresceina e l’ICGA.
Riclassificazione delle forme di malattia: Sta progredendo una comprensione integrata come ‘spettro della corioretinite placoide’ che include la maculopatia placoide persistente e la corioretinite placoide implacabile 4). Mirza e Jampol (2012) hanno organizzato le caratteristiche della corioretinite placoide implacabile come entità patologica indipendente e hanno discusso la sua continuità con l’APMPPE 6).
Testi I, Modugno RL, Pavesio C. Multimodal imaging supporting the pathophysiology of white dot syndromes. Journal of ophthalmic inflammation and infection. 2021;11(1):32. doi:10.1186/s12348-021-00261-3. PMID:34529201; PMCID:PMC8446150.
Claudio Furino, Zaid Shalchi, Maria Oliva Grassi, Joao N. Cardoso, Pearse A. Keane, Alfredo Niro, Maria Vittoria Cicinelli, Michele Reibaldi, et al. OCT Angiography in Acute Posterior Multifocal Placoid Pigment Epitheliopathy. Ophthalmic Surg Lasers Imaging Retina. 2019;50(7):428-436. doi:10.3928/23258160-20190703-04.
Souka AA, Hillenkamp J, Gora F, Gabel VP, Framme C.. Correlation between optical coherence tomography and autofluorescence in acute posterior multifocal placoid pigment epitheliopathy. Graefes Arch Clin Exp Ophthalmol. 2006;244(10):1219-1223. doi:10.1007/s00417-006-0343-1. PMID:16639621.
N P Jones. Acute posterior multifocal placoid pigment epitheliopathy. British Journal of Ophthalmology. 1995;79(4):384-389. doi:10.1136/bjo.79.4.384.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.