Die akute posteriore multifokale plakoide Pigmentepitheliopathie (APMPPE) ist eine bilaterale akute entzündliche Erkrankung, die erstmals 1968 von Gass beschrieben wurde 10). Sie ist gekennzeichnet durch multiple scheibenförmige weiße Flecken in den tiefen Netzhautschichten bis zur Ebene des retinalen Pigmentepithels (RPE) am hinteren Pol beider Augen.
In der Klassifikation der klinischen Leitlinien für Uveitis (Jpn J Ophthalmol 2019;123(6):635-696) wird sie als nicht-infektiöse, fundusläsionsdominierte posteriore Uveitis eingeordnet, mit den klinischen Merkmalen Bilateralität, akuter Beginn und vorübergehender Verlauf 1).
Als Pathomechanismus wird eine obliterative Vaskulitis aufgrund einer verzögerten Überempfindlichkeitsreaktion (Typ-IV-Allergie) in den afferenten Arteriolen der Choriokapillaris angenommen. Virusinfektionen (Influenza, Mumps usw.) werden als Auslöser vermutet, die Details sind jedoch unbekannt.
Das bevorzugte Alter ist 20–30 Jahre (Durchschnitt 25 Jahre), ohne Geschlechtsunterschied 1). Häufig beidseitig. Bei mehr als der Hälfte der Fälle werden vorausgehende grippeähnliche Symptome berichtet 1). Selten wurde ein Zusammenhang mit systemischer Vaskulitis, Nephropathie, Meningoenzephalitis oder Thyreoiditis beschrieben 1).
QTritt APMPPE nur bei jungen Menschen auf?
A
Das bevorzugte Alter sind junge Erwachsene unter 40 Jahren; das Auftreten bei älteren Menschen oder Kindern ist selten. Wenn nach dem 40. Lebensjahr ähnliche Läsionen auftreten, ist die Abgrenzung zur serpiginösen Chorioiditis (geografische Chorioiditis) wichtig.
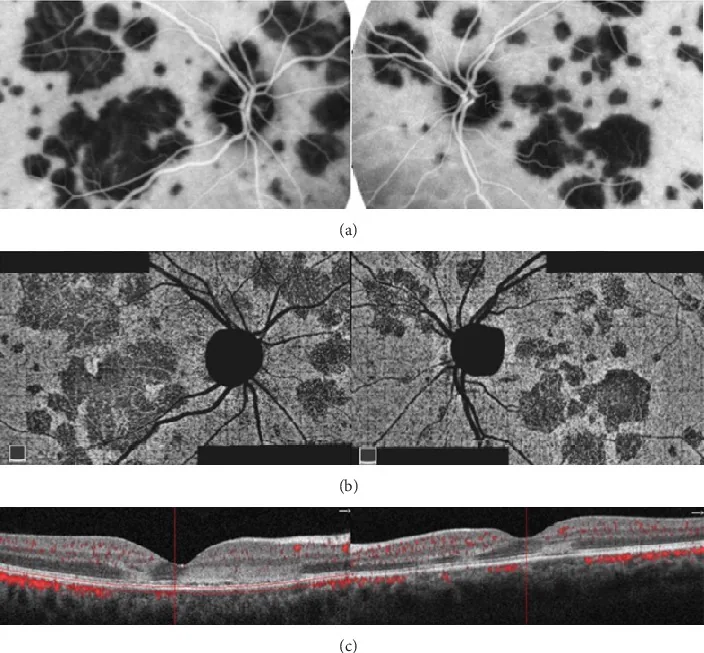
Oliveira MA, et al. Management of Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE): Insights from Multimodal Imaging with OCTA. Case Rep Ophthalmol Med. 2020. Figure 5. PMCID: PMC7094199. License: CC BY.
Am hinteren Pol beider Augen zeigen sich multiple landkartenartige und fleckförmige Läsionen, die in der ICGA hypofluoreszent und in der OCTA als korrespondierende Bereiche verminderter Durchblutung der Choriokapillaris erscheinen. Im OCT zeigen sich hyperreflektive Veränderungen von der äußeren plexiformen Schicht bis zum RPE, was die typischen Befunde der APMPPE darstellt.
Am hinteren Pol beider Augen treten multiple grau-weiße (cremefarbene bis weiße) scheibenförmige weiße Flecken in den tiefen Netzhautschichten bis zur RPE-Ebene auf. Die Läsionen sind scharf begrenzt, nahezu einheitlich in der Größe (1/4 bis 1/2 Papillendurchmesser), treten in geringer bis großer Anzahl auf und zeigen eine geringe Tendenz zur Konfluenz oder Ausdehnung. Leichte Rötung und Schwellung der Papille sowie leichte Glaskörperzellen können beobachtet werden 1). Ein Unterscheidungsmerkmal ist, dass sie größer sind als die weißen Flecken des MEWDS.
Akutphase-Befunde
Fundus weiße Flecken: Multiple scharf begrenzte grau-weiße scheibenförmige Flecken von 1/4 bis 1/2 Papillendurchmesser am hinteren Pol. Ischämisches Ödem und Trübung der äußeren Netzhaut und des RPE aufgrund des Verschlusses der Choriokapillaris.
Äußere Netzhautschädigung: Im OCT zeigt sich eine hyperreflektive Störung der Ellipsoidzone (IS/OS-Linie) 1). Die Zerstörung der Ellipsoidzone führt zu subjektiven Symptomen 2).
FAF (Fundusautofluoreszenz): In der Akutphase erscheinen die Läsionen hypo- oder hyperautofluoreszent. Die Befunde von OCT und Autofluoreszenz korrelieren eng 9).
Remissionsphase-Befunde
Rückbildung der weißen Flecken: Die weißen Flecken bilden sich innerhalb von 7–12 Tagen von der Mitte her zurück und verschwinden unter Hinterlassung leichter Depigmentierungsveränderungen.
OCT-Erholung: Die Ellipsoidzone kann sich innerhalb weniger Monate erholen, jedoch kann eine Ausdünnung der Photorezeptorschicht bestehen bleiben 1).
Aderhaut-Netzhaut-Atrophie: In einigen Fällen bleiben leichte Aderhaut-Netzhaut-Atrophieflecken bestehen1).
QVerschwinden die weißen Flecken der APMPPE?
A
In den meisten Fällen bilden sich die weißen Flecken 7–12 Tage nach Beginn zurück und verschwinden unter Hinterlassung einer Depigmentierung. Die Sehprognose ist in der Regel gut und Rückfälle sind selten. Bei Übergang in eine geografische Chorioretinopathie (serpiginöse Choroiditis) ist jedoch eine Progression zu beobachten, sodass regelmäßige Nachkontrollen erforderlich sind.
Es wird angenommen, dass eine obstruktive Vaskulitis aufgrund einer verzögerten Überempfindlichkeitsreaktion (Typ-IV-Allergie) an den zuführenden Arteriolen der Choriokapillaris die Ursache ist. Die OCT-A (OCT-Angiographie) zeigt in akuten Läsionen einen Flow Void (fehlendes Blutflusssignal) auf Höhe der Choriokapillaris, was die Ischämie der Choriokapillaris als Kern der Pathologie unterstützt3). Klufas et al. berichteten, dass die OCT-A bei drei Erkrankungen der placoiden Chorioretinitis (APMPPE, PPM, RPC) den Choriokapillaris-Flow-Void mit hoher Übereinstimmung mit FA und ICGA nachweist4).
Es gibt Fälle, die nach grippeähnlichen Symptomen (Influenza, Mumps usw.), Infektionen der oberen Atemwege oder Impfungen auftreten.
Es wurden Assoziationen mit systemischer Vaskulitis (zerebrale Vaskulitis), Nephropathie, Polyarteriitis nodosa und Thyreoiditis berichtet1).
QBesteht bei APMPPE ein Schlaganfallrisiko?
A
Obwohl selten, wurden Fälle von APMPPE mit begleitender ZNS-Vaskulitis berichtet. Bei Auftreten neurologischer Symptome (starke Kopfschmerzen, Bewusstseinsveränderungen, Lähmungen) sind eine notfallmäßige MRT und MRA des Gehirns sowie eine Zusammenarbeit mit der Neurologie erforderlich.
Die Diagnose der APMPPE basiert auf charakteristischen Fundusbefunden und Fluoreszenzangiographie-Befunden (FA, ICGA). Die multimodale Bildgebung ermöglicht eine mehrdimensionale Beurteilung der Pathologie 2).
Das in der FA beobachtete „Fluoreszenz-Umkehrphänomen“ (frühe Hypofluoreszenz → späte Hyperfluoreszenz) ist der charakteristischste Befund der APMPPE. Es wird interpretiert als frühe Behinderung des Farbstoffeinstroms in die Choriokapillaris (aufgrund einer okklusiven Vaskulitis) und späte Hyperfluoreszenz durch Exsudation aus der Umgebung 1). In der ICGA zeigt sich von der frühen bis zur späten Phase durchgehend eine Hypofluoreszenz, die die Choriokapillaris-Ischämie direkter widerspiegelt 5).
OCT-A ist eine nicht-invasive Bildgebungsmodalität zur Beurteilung von Durchblutungsstörungen der Choriokapillaris bei APMPPE. Furino et al. berichteten, dass OCT-A bei akuten APMPPE-Läsionen mit hoher Übereinstimmung mit FA und ICGAChoriokapillaris-Flow-Voids darstellen kann 8). In der Erholungsphase wurde eine Verkleinerung der Flow-Voids zusammen mit einer Verbesserung der Sehfunktion beobachtet, was es zu einem nützlichen Biomarker für die Aktivitätsüberwachung macht.
Multiple evaneszente weiße Punkte-Syndrom (MEWDS): Junge Frau, ausgedehnt bis zum Äquator, frühe Hyperfluoreszenz in der FA1)
Persistierende plakoide Makulopathie (PPM) / relentlose plakoide Chorioretinitis (RPC): Der APMPPE ähnliche Form, es gibt Diskussionen über eine Reklassifikation im „plakoiden Chorioretinitis-Spektrum“ 4)7)
QWie unterscheidet man APMPPE von MEWDS?
A
Drei Schlüsselpunkte sind die Läsionsgröße, FA-Befunde und Bilateraliät. Die weißen Flecken der APMPPE sind größer als bei MEWDS (1/4 bis 1/2 Papillendurchmesser), häufig bilateral und zeigen in der FA ein Umkehrphänomen (frühe Hypofluoreszenz, späte Hyperfluoreszenz). MEWDS ist oft unilateral, die Flecken sind kleiner und ausgedehnter, und in der FA zeigt sich von Anfang an eine Hyperfluoreszenz 1).
Die Tendenz zur spontanen Rückbildung ist stark, und die Heilung erfolgt oft ohne spezielle Behandlung 1). Bei leichten Fällen ist die Beobachtung die Grundlage. Auch der Langzeitverlaufsbericht von Jones (1995) zeigt, dass viele Fälle letztendlich eine gute Sehkraft wiedererlangten 10).
Indikationen für eine entzündungshemmende Therapie
Bei deutlicher Sehverschlechterung, Komplikation durch Papillitis oder Läsionen in der Nähe der Makula ist Folgendes in Betracht zu ziehen.
Prednisolon-Tabletten (5 mg): Beginn mit 30 mg/Tag, dann über 2 Wochen bis 1 Monat ausschleichen
Carnaculin-Tabletten (Kallidinogenase 25-50 Einheiten): 3 Tabletten in 3 Einzeldosen (als Begleitmedikation zur Verbesserung der choroidalen Mikrozirkulation)
Bei Übergang in eine geografische Choroidopathie oder Komplikation durch ZNS-Vaskulitis ist eine Steroid-Pulstherapie (Methylprednisolon hochdosiert i.v.) oder der Einsatz von Immunsuppressiva zu erwägen 1). Bei Auftreten einer choroidalen Neovaskularisation (CNV) ist eine intravitreale Anti-VEGF-Injektion in Betracht zu ziehen, jedoch handelt es sich um eine seltene Komplikation. Bei Übergang in eine persistierende plakoide Makulopathie (PPM) kann eine Langzeitbehandlung erforderlich sein, wie Kolomeyer und Brucker in ihrem systematischen Review betonen 7).
QIst eine Steroidbehandlung immer notwendig?
A
Bei leichten Fällen heilt die Erkrankung allein durch Beobachtung spontan aus. Eine Steroidtherapie wird in Betracht gezogen bei deutlicher Sehverschlechterung, starker Entzündung in der Nähe der Papille oder begleitenden ZNS-Symptomen. Auch die Leitlinie zur Uveitis-Behandlung besagt, dass die spontane Heilung ohne spezielle Behandlung die Grundlage ist 1).
Die Pathologie der APMPPE wird als ischämische Veränderung verstanden, die auf den Verschluss der Choriokapillaris zentriert ist.
Es wird angenommen, dass eine durch eine verzögerte Überempfindlichkeitsreaktion (Typ-IV-Allergie) in den zuführenden Arteriolen der Choriokapillaris verursachte obliterative Vaskulitis die Ursache ist. Verschluss → ischämische Veränderungen des RPE und der äußeren Netzhautschichten (Ödem, Trübung) → Bildung scheibenförmiger weißer Flecken. Mrejen et al. zeigten, dass die choroidalen Läsionen tiefer reichen, was auf eine mögliche Beteiligung nicht nur der Choriokapillaris, sondern auch der mittleren und großen Gefäße hindeutet 3).
In der OCT-A werden Flow-Voids auf der Ebene der inneren Aderhaut beobachtet, und es wurde gezeigt, dass eine Ischämie der Choriokapillaris durchgängig als pathophysiologische Grundlage der Krankheitsgruppe des „Placoid-Disorder-Spektrums“ (einschließlich persistierender Plakoid-Makulopathie und unerbittlicher Plakoid-Chorioretinitis) ähnlich der APMPPE vorhanden ist 4). Klufas et al. berichteten, dass die drei Krankheiten ein gemeinsames Muster von Choriokapillaris-Flow-Voids in der OCT-A aufweisen, was das Konzept des Plakoid-Chorioretinitis-Spektrums unterstützt.
Mechanismus des Fluoreszenz-Umkehrphänomens: In der frühen Kontrastmittelphase ist der Farbstoffeinstrom in die Choriokapillaris behindert, was zu einer Hypofluoreszenz führt; in der späten Phase tritt Farbstoff aus dem umliegenden normalen Gewebe aus und wandelt sich in eine Hyperfluoreszenz um. Dass die ICGA durchgehend eine Hypofluoreszenz zeigt, spiegelt ebenfalls die obliterativen Veränderungen der Choriokapillaris wider 5). Die FAF ermöglicht eine nicht-invasive Beurteilung der Verteilung von RPE-Schäden und zeigt eine hohe Übereinstimmung mit den OCT-Befunden 9).
APMPPE nach COVID-19-Infektion/-Impfung: Es wurden Fälle von akuten APMPPE-ähnlichen Läsionen nach einer Infektion berichtet, und die Beziehung zur virusinduzierten Immunantwort wird beachtet.
Komplikation einer zerebralen Vaskulitis: Die Empfehlung für eine zerebrale MRT/MRA bei Patienten mit neurologischen Symptomen nimmt zu. Mehrere Fälle von APMPPE mit Schlaganfall oder zerebraler Vaskulitis wurden berichtet, was die Bedeutung einer systemischen Überwachung zusätzlich zu den Augensymptomen unterstreicht 6).
Fortschritte in der multimodalen Bildgebung: Die Überwachung von Choriokapillaris-Flow-Voids mittels OCT-A könnte ein Biomarker für die Aktivitätsbeurteilung werden 8). Da sie nicht-invasiv und wiederholbar ist, wird sie zu einer wichtigen Modalität, die FA und ICGA ergänzt.
Reklassifikation der Krankheitsformen: Ein integriertes Verständnis als „Plakoid-Chorioretinitis-Spektrum“ einschließlich persistierender Plakoid-Makulopathie und unerbittlicher Plakoid-Chorioretinitis schreitet voran 4). Mirza und Jampol (2012) ordneten die Merkmale der unerbittlichen Plakoid-Chorioretinitis als eigenständige Krankheitsentität und diskutierten ihre Kontinuität mit der APMPPE 6).
Testi I, Modugno RL, Pavesio C. Multimodal imaging supporting the pathophysiology of white dot syndromes. Journal of ophthalmic inflammation and infection. 2021;11(1):32. doi:10.1186/s12348-021-00261-3. PMID:34529201; PMCID:PMC8446150.
Claudio Furino, Zaid Shalchi, Maria Oliva Grassi, Joao N. Cardoso, Pearse A. Keane, Alfredo Niro, Maria Vittoria Cicinelli, Michele Reibaldi, et al. OCT Angiography in Acute Posterior Multifocal Placoid Pigment Epitheliopathy. Ophthalmic Surg Lasers Imaging Retina. 2019;50(7):428-436. doi:10.3928/23258160-20190703-04.
Souka AA, Hillenkamp J, Gora F, Gabel VP, Framme C.. Correlation between optical coherence tomography and autofluorescence in acute posterior multifocal placoid pigment epitheliopathy. Graefes Arch Clin Exp Ophthalmol. 2006;244(10):1219-1223. doi:10.1007/s00417-006-0343-1. PMID:16639621.
N P Jones. Acute posterior multifocal placoid pigment epitheliopathy. British Journal of Ophthalmology. 1995;79(4):384-389. doi:10.1136/bjo.79.4.384.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.