Die idiopathische multifokale Choroiditis (IMFC) ist eine spontan auftretende entzündliche Erkrankung mit multiplen Läsionen in der Netzhaut und Aderhaut. Sie verläuft in wiederkehrenden Entzündungsschüben und tritt beidseitig, gleichzeitig oder zeitlich versetzt auf.
1984 berichteten Deutsch und Tessler über 28 Fälle als „Pseudo-POHS“. 1986 beschrieben Morgan und Shatz 11 Fälle als „rezidivierende multifokale Choroiditis“ und wiesen auf die Vitritis als charakteristisches Merkmal hin, das bei POHS-Patienten fehlt. Die IMFC ist eine eigenständige Erkrankung, die von der PIC (punktförmige innere Choroidopathie), der multifokalen Choroiditis mit Panuveitis (MFCwP) und dem Syndrom der progressiven subretinalen Fibrose mit Uveitis abgegrenzt wird.
Sowohl MFC als auch PIC sind Subtypen des White-Dot-Syndroms (WDS), einer Gruppe entzündlicher Erkrankungen, die vor allem die äußere Netzhaut, die Choriokapillaris und die Aderhaut betreffen 1). Es wurde auch vermutet, dass MFC und PIC möglicherweise dasselbe Krankheitsspektrum darstellen 1).
QWas ist der Unterschied zwischen POHS und IMFC?
A
Der Hauptunterschied ist das Vorhandensein oder Fehlen einer Vitritis. POHS geht nicht mit einer Vitritis einher, während bei IMFC in der Regel eine ein- oder beidseitige Vitritis vorliegt. Zudem kann bei IMFC auch eine leichte Entzündung der Vorderkammer auftreten.
Papasavvas I, et al. Idiopathic multifocal choroiditis (MFC): aggressive and prolonged therapy with multiple immunosuppressive agents is needed to halt the progression of active disease. An offbeat review and a case series. J Ophthalmic Inflamm Infect. 2022. Figure 2. PMCID: PMC8743334. License: CC BY.
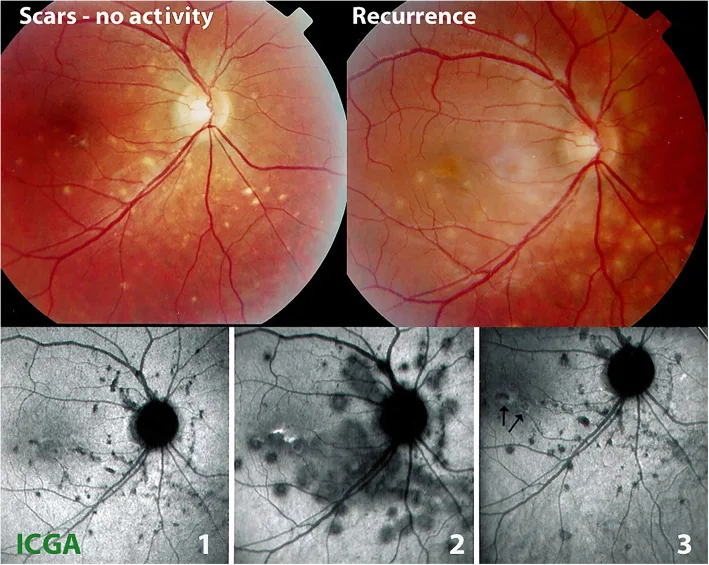
Auf der Fundusfotografie (obere Reihe) einer multifokalen Choroiditis (MFC) sind links in der Ruhephase gelbe ausgestanzte Läsionen und rechts bei einem Rezidiv neue ödematöse Läsionen zu erkennen. Die ICGA-Bilder (untere Reihe) zeigen den Verlauf in der Ruhephase (1), der aktiven Phase (2) und nach Behandlung (3). Sie entsprechen den im Abschnitt „2. Hauptsymptome und klinische Befunde“ beschriebenen choroidalen Atrophieläsionen.
Die Ätiologie der IMFC ist unbekannt. Es wird vermutet, dass eine vorausgehende Infektion eine Immunreaktion auslöst, aber ein spezifischer Erreger wurde nicht identifiziert.
Epidemiologische Merkmale:
Häufiger bei weißen Frauen.
Durchschnittliches Erkrankungsalter 30 Jahre (Spanne: 6–69 Jahre).
Die meisten Patienten sind kurzsichtig.
Betrifft gesunde Personen ohne bekannte systemische oder Augenerkrankungen.
Genetische Veranlagung:
Die idiopathische multifokale Choroiditis ist mit IL-10- und Tumornekrosefaktor (TNF)-Haplotypen assoziiert.
Pathogenese-Hypothese:
Die entzündlichen Läsionen beginnen auf der Ebene des retinalen Pigmentepithels und der Choriokapillaris. Ein exogenes Antigen kann eine Antigensensibilisierung in den Photorezeptoren und dem retinalen Pigmentepithel auslösen, was die Integrität der Bruch-Membran beeinträchtigen kann. Dadurch entsteht Raum für die Entwicklung einer choroidalen Neovaskularisationsmembran, die bei bis zu 60 % der Patienten auftreten kann.
Die idiopathische multifokale Choroiditis ist eine klinische Diagnose und eine Ausschlussdiagnose. Der Ausschluss von infektiösen, malignen und systemischen Erkrankungen ist zwingend erforderlich.
OCT-Angiographie (OCT-A): Bereiche verminderter Durchblutung auf Höhe der Choriokapillaris (entsprechend aktiven Entzündungsläsionen)1).
Fluoreszenzangiographie-Befunde:
Akute Entzündungsläsionen zeigen eine späte Anfärbung; bei Verdacht auf choroidale Neovaskularisation erfolgt die Beurteilung durch Kombination von Leckagemuster und OCT-Befunden1).
In tuberkuloseendemischen Regionen (z. B. Indien) können bis zu 40 % der multifokalen Choroiditiden mit einer okulären Tuberkulose assoziiert sein2) und im OCT als entzündliche Läsionen unter dem RPE beobachtet werden2).
QWie wird die idiopathische multifokale Choroiditis von der Tuberkulose unterschieden?
A
Die multimodale Bildgebung ist für die Differentialdiagnose nützlich. Bei der tuberkulösen multifokalen Choroiditis kann die optische Kohärenztomographie einen Riss der äußeren Grenzmembran oder einen lokalisierten Verlust der Ellipsoidzone in den entzündlichen Läsionen unter dem retinalen Pigmentepithel zeigen 2). Die Beurteilung erfolgt durch Kombination des QuantiFERON-TB-Gold-Tests und der Bildgebung.
Die Behandlung wird basierend auf dem Schweregrad der Entzündung, aktiven Läsionen, Komplikationen und Sehverschlechterung ausgewählt. Ein zystoides Makulaödem, eine dichte Vitritis oder das Auftreten einer choroidalen Neovaskularisationsmembran sind Behandlungsindikationen.
Orale Steroide: Erstlinientherapie. Beginn mit hoher bis mittlerer Dosis, dann schrittweise Reduktion entsprechend dem Abklingen der Entzündung.
Lokale Steroide:
Triamcinolonacetonid (Kenacort-A): subtenonale oder intravitreale Injektion. Wirkdauer 2–3 Monate.
Dexamethason-Intravitrealimplantat (Ozurdex®): indiziert für nichtinfektiöse Uveitis des hinteren Augenabschnitts. Wirkdauer 3–4 Monate. Erfordert eine Augeninnendruckmessung innerhalb von 30 Minuten nach der Verabreichung und eine Spaltlampenuntersuchung nach 2–7 Tagen.
Fluocinolonacetonid-Mikroimplantat (Iluvien®): kontinuierliche Freisetzung niedrig dosierter Steroide für bis zu 3 Jahre.
Bei schweren oder therapierefraktären Fällen sollten Immunmodulatoren wie Antimetaboliten, Biologika oder T-Zell-Inhibitoren in Betracht gezogen werden.
Laut einer Umfrage einer internationalen Forschungsgruppe werden Immunmodulatoren wie Methotrexat und Adalimumab häufig bei Erkrankungen des multifokalen Choroiditis-PIC-Spektrums eingesetzt 3).
Es wird angenommen, dass die Entzündungsherde auf der Ebene des retinalen Pigmentepithels und der Choriokapillaris beginnen und dass eine Antigensensibilisierung im retinalen Pigmentepithel durch fremde Antigene erfolgt.
Die idiopathische multifokale Choroiditis und die punktierte innere Choroidopathie betreffen beide die äußere Netzhaut, die Choriokapillaris und die Aderhaut und könnten zum selben Krankheitsspektrum gehören 1). Die multifokale Choroiditis ist eine chronische, bilaterale, rezidivierende entzündliche Erkrankung, die durch hintere Läsionen mit anteriorer Uveitis und Glaskörperentzündung gekennzeichnet ist, während sich die punktierte innere Choroidopathie durch das Fehlen von Glaskörperentzündung und anterioren Entzündungszeichen unterscheidet 1).
OCT-Angiographie-Befunde zeigen deutliche Durchblutungsminderungen auf der Ebene der Choriokapillaris, die aktiven Entzündungsherden entsprechen, was die Annahme stützt, dass Veränderungen der äußeren Netzhaut sekundäre Folgen einer primären Aderhautbeteiligung sind 1).
Die Entzündung beeinträchtigt die Integrität der Bruch-Membran, was als Ausgangspunkt für die Entwicklung einer choroidalen Neovaskularisationsmembran dient. Bei der idiopathischen multifokalen Choroiditis kann bei bis zu 60 % der Patienten eine choroidale Neovaskularisation auftreten.
Als genetische Faktoren wurde eine Assoziation mit Haplotypen von IL-10 und Tumornekrosefaktor nachgewiesen, der genaue Mechanismus bleibt jedoch weiterhin unklar.
7. Aktuelle Forschung und Zukunftsperspektiven (Forschungsstadium)
Die OCT-Angiographie zeigt eine deutliche Durchblutungsminderung in der Choriokapillaris von Läsionen der punktierten inneren Choroidopathie und multifokalen Choroiditis, die aktiven Entzündungsherden entspricht 1). Sie wird auch zur Überwachung von Aderhautgefäßveränderungen nach der Behandlung eingesetzt.
Es wurde gezeigt, dass die OCT-Angiographie Bereiche mit verminderter Durchblutung in der Choriokapillaris darstellen kann, indem sie sie mit Erhebungen des retinalen Pigmentepithels in der optischen Kohärenztomographie und hypofluoreszenten Flecken in der Indocyaningrün-Angiographie korreliert, was das pathophysiologische Verständnis vertieft 1).
Die multimodale Bildgebung hat die Ansicht hervorgebracht, dass multifokale Choroiditis mit Panuveitis und punktuelle innere Choroidopathie als ein Spektrum betrachtet werden können. Es werden genauere Klassifikationen und personalisierte Behandlungen entwickelt. 1)
Testi I, Modugno RL, Pavesio C. Multimodal imaging supporting the pathophysiology of white dot syndromes. Journal of ophthalmic inflammation and infection. 2021;11(1):32. doi:10.1186/s12348-021-00261-3. PMID:34529201; PMCID:PMC8446150.
Kaza H, Gala JM, Rani PK. Subfoveal retinal pigment epithelium inflammatory lesion presenting as a sign of reactivation of tubercular multifocal choroiditis. BMJ case reports. 2021;14(5). doi:10.1136/bcr-2020-240280. PMID:34031072; PMCID:PMC8149327.
Branford JA, et al. Practice patterns of systemic immunomodulatory drug treatment for non-infectious uveitis: an international study. Br J Ophthalmol. 2025;109:482-489.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.