A doença de Behçet é uma doença inflamatória sistêmica refratária de causa desconhecida que apresenta quatro sintomas principais: úlceras aftosas orais, lesões oculares, úlceras genitais e lesões cutâneas. A inflamação é aguda e transitória, mas a característica da doença é a recorrência repetida. Ocorre em adultos jovens de 20 a 50 anos, com uma proporção entre homens e mulheres de aproximadamente 1:1. A uveíte ocorre em cerca de 70% dos pacientes do sexo masculino e em cerca de 45% das pacientes do sexo feminino, sendo mais grave em homens jovens.
É frequente ao longo da Rota da Seda, desde o Mediterrâneo até a China e o Japão. Cerca de 50% dos pacientes são HLA-B51 positivos (15% na população geral)5), e HLA-A26 (A*2601) também foi relatado como um alelo de risco independente. Acredita-se que fatores imunogenéticos e ambientais estejam envolvidos no desenvolvimento da doença. Antigamente, a doença tinha uma alta taxa de cegueira, mas atualmente, com o avanço dos imunossupressores e agentes biológicos, a taxa de cegueira diminuiu.
A doença de Behçet está registrada como doença rara designada 568).
Características da doença
Idade de início: Adultos jovens de 20 a 50 anos
Diferença de sexo: Proporção homem:mulher aproximadamente 1:1. As lesões oculares são mais graves em homens jovens.
Distribuição geográfica: Frequente ao longo da Rota da Seda (Mediterrâneo, Oriente Médio, Leste Asiático)
Fundo genético: HLA-B51 positivo (cerca de 50% dos pacientes), HLA-A26 também é um risco independente5)
4 sintomas principais
Úlcera aftosa oral: úlcera recorrente dolorosa. Frequentemente é o sintoma inicial
A proporção da doença de Behçet entre todas as uveítes tem diminuído ao longo dos anos. Em um estudo epidemiológico de 2002, era de 6,2% (3º lugar), mas em 2009 caiu para 3,9% (6º lugar)5). Além da redução no número de pacientes, também foi relatada uma tendência a formas mais leves da doença4).
QO que é a doença de Behçet?
A
A doença de Behçet é uma doença inflamatória sistêmica recidivante que apresenta quatro sintomas principais: úlceras aftosas orais, sintomas oculares, úlceras genitais e sintomas cutâneos. A causa é desconhecida, mas suspeita-se de um mecanismo autoimune. É mais comum em regiões ao longo da Rota da Seda, e acredita-se que fatores genéticos (HLA-B51) e ambientais estejam envolvidos. As lesões oculares são particularmente importantes e podem levar à cegueira se não tratadas adequadamente.
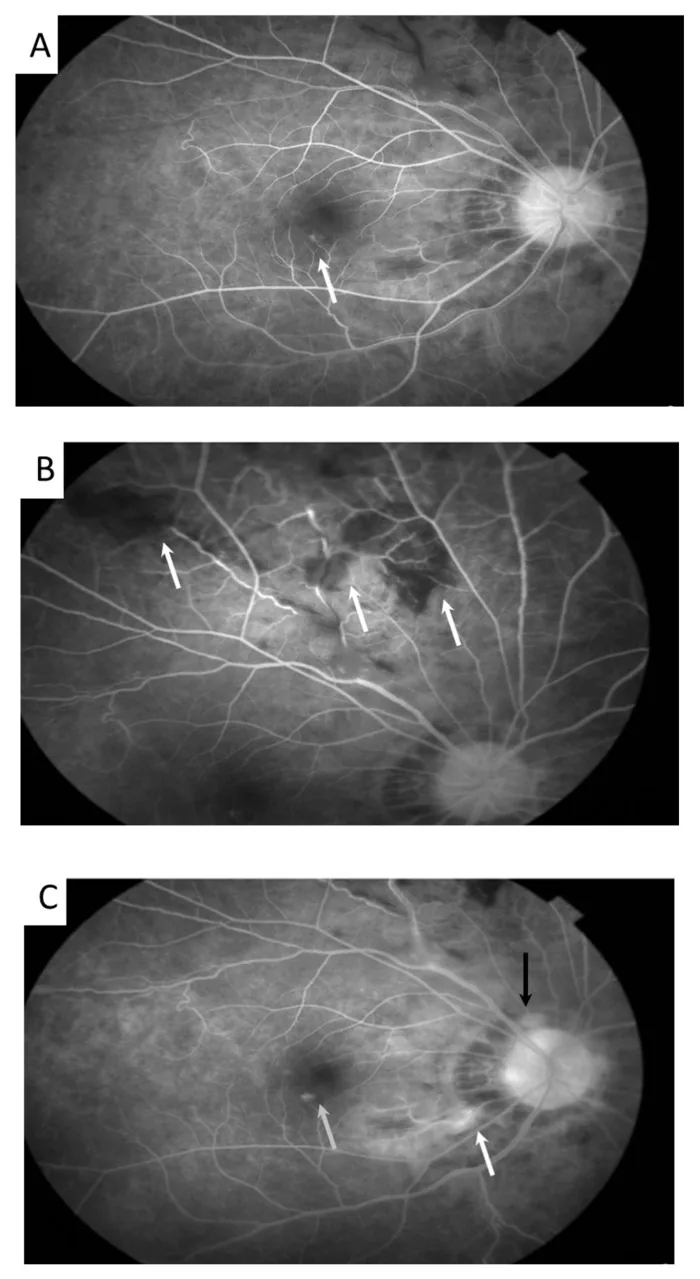
Meng PP, et al. Use of Ultra-Widefield Fluorescein Angiography to Guide the Treatment to Idiopathic Retinal Vasculitis, Aneurysms, and Neuroretinitis-Case Report and Literature Review. Medicina (Kaunas). 2022. Figure 2. PMCID: PMC9611749. License: CC BY.
Na imagem de autofluorescência, observam-se dilatações aneurismáticas das arteríolas retinianas (setas) e tortuosidade venosa. Corresponde aos achados de vasculite retiniana discutidos na seção “2. Principais sintomas e achados clínicos”.
Como sintomas subjetivos oculares, durante a fase de crise inflamatória podem ocorrer visão turva, diminuição da acuidade visual e moscas volantes. A inflamação geralmente se repete em momentos diferentes em cada olho, e cerca de 90% dos casos tornam-se bilaterais. Em casos graves, a frequência das crises pode ser de várias vezes ao mês, e mesmo em casos leves, cerca de uma crise por ano pode persistir por vários anos a mais de uma década. Pode ocorrer retinite recorrente limitada à mácula, causando perda irreversível da acuidade visual.
Nos sintomas sistêmicos, observam-se principalmente úlceras aftosas orais (cerca de 90%), sintomas cutâneos (cerca de 75%) e úlceras genitais (cerca de 50%). As úlceras aftosas orais ocorrem frequentemente na língua, mucosa jugal, lábios e gengivas, sendo úlceras dolorosas com vermelhidão ao redor. São quase sempre presentes na doença e frequentemente o primeiro sintoma. Curam-se sem cicatriz em até 10 dias, mas recorrem.
Como sintomas secundários, há artrite sem deformidade ou rigidez, epididimite, doença de Behçet gastrointestinal (úlcera ileocecal), doença de Behçet vascular (vasculite) e doença de Behçet neurológica (meningoencefalite). Como os sintomas extraoculares e os achados oculares nem sempre estão correlacionados, é importante avaliar o estado geral além da observação oftalmológica3).
Os seguintes dois sintomas oculares se repetem de forma paroxística.
(1) Iridociclite aguda que pode ser acompanhada de hipópio.
(2) Opacidade vítrea difusa, coriorretinite e retinovasculite.
Achados na fase precoce e ativa do ataque ocular: Inflamação ocular paroxística. Precipitados ceráticos finos, células inflamatórias na câmara anterior e hipópio. Geralmente não há exsudação de fibrina na câmara anterior. Não forma nódulos irianos ou goniosinequias (não granulomatoso). No segmento posterior, é característico que a opacidade vítrea, exsudatos retinianos e hemorragias desapareçam relativamente rápido (1 a 2 semanas)3).
Achados tardios3): Atrofia coriorretiniana, esbranquiçamento dos vasos retinianos e palidez do disco óptico, levando a grave diminuição da função visual.
O hipópio na doença de Behçet é fluido devido à infiltração predominante de neutrófilos, formando um nível horizontal nítido. É característico que se mova facilmente com mudanças de posição, diferindo do hipópio viscoso da uveíte associada ao HLA-B273). Os precipitados ceráticos são finos e apresentam aspecto não granulomatoso.
Durante a exacerbação aguda da uveíte posterior, observam-se opacidade vítrea, retinovasculite, exsudatos retinianos brancos e hemorragias. Os exsudatos brancos são devidos à infiltração de leucócitos e edema das fibras nervosas ópticas por isquemia, e têm a característica de desaparecer relativamente rápido (cerca de uma semana). A vasculite oclusiva pode causar hemorragias semelhantes à oclusão de ramo venoso retiniano.
Na angiografia fluoresceínica, mesmo na ausência de crises oculares, frequentemente observa-se um vazamento fluorescente intenso e difuso (vazamento em forma de samambaia) dos capilares retinianos, considerado característico desta doença.
BOS24 (Escore de Crise Ocular da Doença de Behçet 24) 6)
Escala que quantifica o grau de inflamação em cada crise ocular. Seis itens: inflamação na câmara anterior (máx. 4 pontos), opacidade vítrea (máx. 4), lesões retinianas periféricas (máx. 8), lesões retinianas do polo posterior (máx. 4), lesão foveal (máx. 2) e lesão do nervo óptico (máx. 2), totalizando 24 pontos. Utilizada para avaliação objetiva da gravidade da crise e determinação da eficácia terapêutica 6).
Além disso, a inflamação intraocular pode complicar com catarata secundária e glaucoma secundário, causando deficiência visual.
Achados cutâneos: eritema nodoso predominante nas pernas, tromboflebite subcutânea, erupções foliculite-like ou acne-like na face, pescoço e costas. Úlceras genitais são aftosas, dolorosas, com bordas nítidas, predominando no escroto e pênis em homens, e nos grandes e pequenos lábios em mulheres.
Em pacientes pediátricos, também foram relatados sintomas oculares graves, como uveíte posterior, vasculite retiniana e papilite 1), com atraso diagnóstico médio de 11,3±8,5 meses em alguns casos 1).
QComo diferenciar o hipópio na doença de Behçet?
A
O hipópio na doença de Behçet é de natureza neutrofílica, portanto “fluido”, formando um nível horizontal nítido e móvel com a mudança de posição. Em contraste, o hipópio na uveíte associada ao HLA-B27 é mais viscoso, não forma nível e tem aparência elevada 3). Essa diferença de característica é um indício diagnóstico.
A causa da doença de Behçet é desconhecida, mas acredita-se que fatores externos, como microrganismos patogênicos (estreptococos), e fatores internos, como predisposição genética e anormalidades imunológicas, estejam envolvidos. Anormalidades na função dos neutrófilos e citocinas, como o TNF-α, desempenham um papel central, resultando em inflamação paroxística e recorrente nas mucosas oral, ocular, pele e genitália.
Fatores de Risco
Descrição
HLA-B51
Positivo em cerca de 50% dos pacientes (15% na população geral). Principal marcador genético 5)
HLA-A26
Relatado como alelo de risco independente 2)
Região/Etnia
Alta frequência ao longo da Rota da Seda (Mediterrâneo, Oriente Médio, Leste Asiático)
Sexo/Idade
Início entre 20 e 50 anos. Homens jovens apresentam mais lesões oculares graves
Em crianças, representa cerca de 1,6 a 7,7% de todos os casos de doença de Behçet2), e a gravidade das lesões oculares em casos HLA-B51 positivos tem sido relatada1).
:::tip Cuidados diários
A colchicina requer uso prolongado para prevenir ataques oculares. Mesmo que os ataques oculares pareçam ter cessado, não interrompa a medicação por conta própria. Além disso, sabe-se que o tratamento com fotocoagulação pode desencadear ataques intensos, por isso é importante discutir completamente com o médico responsável.
:::
O diagnóstico da doença de Behçet é baseado nas diretrizes clínicas do grupo de pesquisa da doença de Behçet do Ministério da Saúde, Trabalho e Bem-Estar (1987, revisão menor em 2016). Para o diagnóstico baseado em sintomas oculares, consulte as Diretrizes Clínicas para Lesões Oculares da Doença de Behçet (2012) 3).
O tipo completo é definido como o aparecimento de todos os 4 sintomas principais durante o curso da doença, e o tipo incompleto como o aparecimento de 3 sintomas principais ou 2 sintomas principais e 2 sintomas secundários (ou sintomas oculares típicos e 1 outro sintoma principal e 2 sintomas secundários).
5 itens de sintomas secundários (revisão menor de 2016)8): (1) artrite sem deformidade ou rigidez, (2) epididimite, (3) lesões gastrointestinais representadas por úlcera ileocecal, (4) lesões vasculares, (5) lesões do sistema nervoso central de gravidade moderada ou superior
Tipo
Requisitos
Tipo completo
Todos os 4 sintomas principais aparecem
Tipo incompleto
3 sintomas principais ou 2 sintomas principais + 2 sintomas secundários
Tipo especial
Tipo intestinal, vascular e neurológico (quando preenche os critérios de forma completa ou incompleta e apresenta lesões especiais)
Os critérios diagnósticos internacionais do International Study Group for Behçet’s Disease (ISG criteria, 1990) 7) também são amplamente utilizados internacionalmente.
Exame com lâmpada de fenda: inflamação da câmara anterior (flare, contagem de células, hipópio). Quantificado pelo escore BOS24
Exame de fundo de olho: confirmação de opacidade vítrea, vasculite retiniana, exsudatos e hemorragias
Angiografia fluoresceínica (FA): vazamento fluorescente em forma de samambaia (detectável mesmo em períodos não agudos)
OCT: avaliação do edema macular cistóide
Exames sistêmicos:
Teste HLA-B51 (auxílio diagnóstico)
Exames de sangue: leucócitos, PCR, VHS (marcadores inflamatórios)
Teste de patergia (pathergy test): eritema e formação de pústulas após punção intradérmica (diagnóstico auxiliar)
Em casos pediátricos, o diagnóstico tende a ser tardio, com relatos de um intervalo médio de 11,3 ± 8,5 meses entre o início dos sintomas oculares e o diagnóstico1).
Uveíte associada ao HLA-B27: hipópio viscoso, de formato irregular com elevação central. Quase sem inflamação do segmento posterior3). Endoftalmite fúngica: forma hipópio e opacidade vítrea, mas é progressiva. Irite diabética: raramente forma hipópio3).
Colchicina oral (não aprovado para esta indicação): 0,5–1,5 mg/dia, geralmente 1 mg/dia dividido em duas doses. Cerca de 60% dos pacientes apresentam melhora parcial3). Efeitos colaterais: sintomas gastrointestinais como diarreia, teratogenicidade. Mesmo após o controle dos ataques oculares, é necessário tratamento prolongado, com exames de sangue regulares (monitoramento de função hepática e renal, granulocitopenia, rabdomiólise).
Ciclosporina (Neoral® 50 mg): 5 mg/kg/dia como referência (dividido em duas doses). Monitoramento com alvo de concentração mínima de 50–200 ng/mL.
:::caution Efeitos colaterais importantes da ciclosporina
A disfunção renal ocorre com alta frequência. Além disso, sabe-se que a doença de Behçet neurológica se manifesta em cerca de 20% dos casos, sendo necessário atenção aos sintomas neurológicos durante o uso prolongado3). A combinação com colchicina apresenta risco de miopatia.
:::
Passo 4: Inibidores de TNF (casos refratários e graves)
Aprovação: Aprovado em 2007 para uveíte retiniana refratária associada à doença de Behçet
Indicação: Uveíte retiniana refratária resistente ao tratamento existente ou quando o uso de imunossupressores é difícil devido a efeitos colaterais sistêmicos
Posologia: 5 mg/kg em infusão intravenosa durante pelo menos 2 horas. Administrar na 2ª e 6ª semanas após a primeira dose, depois a cada 8 semanas. Administrar através de um filtro de membrana de 1,2 μm ou menor. Após a 6ª semana, se não houver reação à infusão, o tempo de infusão pode ser reduzido (desde que a taxa média de infusão não exceda 5 mg/kg por hora)
Falta de resposta primária e secundária: A falta de resposta primária ocorre quando não há efeito desde o início; a falta de resposta secundária ocorre quando o efeito diminui durante o tratamento
Triagem pré-administração (diretrizes para uso de inibidores de TNF)9):
Radiografia de tórax, teste cutâneo de tuberculina; se fortemente positivo, QFT/T-SPOT
Lesão tuberculosa antiga → Isoniazida oral desde 3 semanas antes do início do infliximabe até o 9º mês
HBsAg, HBsAb, HBcAb (monitoramento de reativação do HBV)
HBsAg positivo → consulta obrigatória com hepatologista. Mesmo negativo, se HBsAb/HBcAb positivo (infecção prévia) → medição regular de HBV DNA
Verificação da presença de hepatite C, HTLV-1 e HIV
Doenças de base: insuficiência cardíaca congestiva, doenças desmielinizantes, neoplasias malignas
Contraindicações9): Infecções incluindo tuberculose ativa (infecções micobacterianas atípicas, infecção pelo vírus da hepatite B), insuficiência cardíaca congestiva (NYHA classe III ou superior), neoplasias malignas, doenças desmielinizantes (como esclerose múltipla)
Monitoramento de efeitos colaterais9): Exames de sangue periférico regulares (leucócitos, linfócitos) e exames bioquímicos (incluindo PCR). Atenção ao desenvolvimento de tuberculose e pneumonia por Pneumocystis (radiografia de tórax, TC, β-D-glucana). Atenção à reativação da infecção prévia pelo vírus da hepatite B (HBV-DNA). Reações à infusão (observação durante e por 2 horas após a infusão). Atenção também à hipersensibilidade tardia (mialgia, erupção cutânea, febre, artralgia após 3 ou mais dias da administração).
Critérios para médicos e instituições9): Especialista certificado pela Sociedade Japonesa de Oftalmologia e membro da Sociedade Japonesa de Uveíte, conclusão do e-learning da mesma sociedade. A instituição introdutória deve ser registrada na Sociedade Japonesa de Uveíte. É necessária capacidade de lidar com efeitos colaterais graves, suporte respiratório/infeccioso e colaboração com internistas experientes em inibidores de TNF.
Caso representativo: Homem de 32 anos, HLA-B51 positivo, forma completa. Incontrolável com ciclosporina, prednisolona e colchicina → introdução de infliximabe. 3 crises oculares em ambos os olhos no ano anterior ao início → nenhuma crise no ano seguinte. Acuidade visual corrigida melhorou de 1,2 (OD)/0,7 (OE) para 1,2 (OD)/0,9 (OE). Boa acuidade visual mantida após 3 anos e 6 meses3).
Em alguns casos, a redução ou descontinuação pode desencadear crises inflamatórias oculares graves, piorando o prognóstico visual. Geralmente, o uso prolongado de corticosteroides orais não é recomendado, mas pode ser utilizado por um período muito curto de cerca de 1 semana quando há alterações exsudativas maculares significativas3).
Azatioprina: frequentemente usada no exterior. Pode ser considerada de primeira linha12)
Injeção intravítrea de triancinolona acetonida: efeito supressor de crises enquanto o medicamento estiver no vítreo. Requer injeções repetidas, com atenção aos efeitos colaterais de catarata e aumento da pressão intraocular.
Interferon-alfa-2a: usado principalmente na Europa. Alta eficácia relatada12)
Cuidados com pacientes cirúrgicos (diretrizes para uso de inibidores de TNF)9)
Em cirurgias intraoculares minimamente invasivas, a suspensão do inibidor de TNF não é absolutamente indicada. Em cirurgias extraoculares ou de outros órgãos com maior invasão, considerar a suspensão (meia-vida do infliximabe: aproximadamente 8-9,5 dias; adalimumabe: aproximadamente 14 dias).
Glaucoma secundário: adicionar colírios, medicamentos orais ou intravenosos antiglaucomatosos.
Catarata concomitante: é desejável um período livre de crises de 6 meses ou mais. Cirurgia (com implante de lente intraocular) após controle da inflamação.
Edema macular cistóide: injeção subtenoniana de corticosteroide e intensificação de imunossupressores.
Opacidade vítrea, hemorragia, descolamento tracional da retina: considerar vitrectomia
Fotocoagulação retiniana: realizada para vasculite oclusiva, mas não deve ser feita levianamente, pois pode induzir ataques inflamatórios oculares graves3)
QEm quais pacientes o infliximabe é usado?
A
É usado em casos refratários e graves em que os ataques oculares não são controlados com colchicina ou ciclosporina. Somente médicos especialistas em oftalmologia pela Sociedade Japonesa de Oftalmologia e membros da Sociedade Japonesa de Uveíte, que tenham concluído o e-learning, podem prescrevê-lo9). A administração inicial é feita nas semanas 0, 2 e 6, seguida de manutenção a cada 8 semanas. A triagem para tuberculose e hepatite B é obrigatória antes da administração.
6. Fisiopatologia e mecanismos detalhados de desenvolvimento
Acredita-se que o desenvolvimento da doença de Behçet envolva uma combinação de fatores imunogenéticos e ambientais.
Fatores genéticos: HLA-B51 é o marcador genético mais fortemente associado à doença de Behçet, sendo positivo em cerca de 50% dos pacientes (15% na população geral)5). HLA-B15, B27, B40, B44, B52, B57 e A26 também foram identificados como alelos de risco independentes2).
Mecanismo inflamatório: A disfunção dos neutrófilos desempenha um papel central, e a produção excessiva de citocinas, como TNF-α, desencadeia a resposta inflamatória. Intraocularmente, ocorre vasculite oclusiva, manifestada como aumento da permeabilidade dos capilares retinianos (vazamento fluorescente em forma de samambaia na angiografia fluoresceínica). Durante a fase aguda de exacerbação, formam-se infiltrados de leucócitos (neutrófilos) e exsudatos brancos devido à isquemia.
Características da inflamação não granulomatosa: Iridociclite não granulomatosa na qual as células inflamatórias não formam aglomerados → precipitados ceráticos finos (KP). Esta é uma característica importante para diferenciar da uveíte granulomatosa, como sarcoidose e doença de Harada3).
Recorrência paroxística da inflamação: Com a repetição dos ataques oculares, ocorre atrofia da retina e do nervo óptico, levando a grave comprometimento da função visual. O fato de o hipópio ser neutrofílico e de consistência fluida também reflete o mecanismo inflamatório predominantemente neutrofílico.
Em casos representativos das diretrizes, foi relatada uma evolução favorável ao longo de 3 anos e 6 meses3). A introdução do infliximabe reduz acentuadamente a frequência dos ataques oculares e mantém ou melhora a acuidade visual10). Uma revisão sobre a administração a longo prazo de infliximabe para a doença de Behçet refratária no Japão também demonstrou alta eficácia na supressão de ataques e na manutenção da visão11).
A comparação de pacientes das décadas de 1980 e 1990 (Yoshida 2004) confirmou uma tendência de gravidade mais leve4). A prevalência diminuiu de 6,2% em 2002 para 3,9% em 20095). Paralelamente à disseminação dos agentes biológicos, a incidência de comprometimento visual grave também diminuiu.
Os inibidores de TNF (infliximabe e adalimumabe) demonstraram eficácia em várias doenças, incluindo uveíte não infecciosa associada à doença de Behçet 12). Particularmente em crianças, o tratamento agressivo com infliximabe tem mostrado boa manutenção da acuidade visual1). O adalimumabe está em fase de acúmulo de evidências para a doença de Behçet, e esperam-se resultados de pesquisas futuras 9).
:::danger Aviso de isenção de responsabilidade
Este artigo tem como objetivo fornecer informações médicas e não indica diagnósticos ou tratamentos individuais. As decisões sobre tratamento devem sempre seguir as orientações de um médico especialista. A dosagem e a administração dos medicamentos variam de acordo com a condição de cada paciente; portanto, consulte seu médico para a prescrição real.
:::
Casem Azri, Perrine Dusser, Laura Eid, Emmanuel Barreau, Isabelle Kone-Paut, Charlotte Borocco, Caroline Galeotti, Sami Saad, et al. Ocular involvement in pediatric Behçet’s disease: is it different than in adults? (a short case series and mini review). BMC Ophthalmol. 2023;23(1). doi:10.1186/s12886-023-03197-5.
Xin Yao, Xing-Ning Wang, Jian-Ming Lai. Pediatric Behçet’s disease with cardiac valvular lesions: A case-based review. Science Progress. 2023;106(2). doi:10.1177/00368504231173404.
Yoshida A, Kawashima H, Motoyama Y, Shibui H, Kaburaki T, Shimizu K, Ando K, Hijikata K, Izawa Y, Hayashi K, Numaga J, Fujino Y, Masuda K, Araie M.. Comparison of patients with Behçet’s disease in the 1980s and 1990s. Ophthalmology. 2004;111(4):810-815. doi:10.1016/j.ophtha.2003.07.018. PMID:15051217.
Kaburaki T, Namba K, Sonoda KH, Kezuka T, Keino H, Fukuhara T, Kamoi K, Nakai K, Mizuki N, Ohguro N, Ocular Behçet Disease Research Group of Japan.. Behçet’s disease ocular attack score 24: evaluation of ocular disease activity before and after initiation of infliximab. Jpn J Ophthalmol. 2014;58(2):120-130. doi:10.1007/s10384-013-0294-0. PMID:24482146.
INTERNATIONALSTUDYGROUPFORBEHC. Criteria for diagnosis of Behcet’s disease. The Lancet. 1990;335(8697). doi:10.1016/0140-6736(90)92643-v.
Ohno S, Nakamura S, Hori S, Shimakawa M, Kawashima H, Mochizuki M, Sugita S, Ueno S, Yoshizaki K, Inaba G.. Efficacy, safety, and pharmacokinetics of multiple administration of infliximab in Behçet’s disease with refractory uveoretinitis. J Rheumatol. 2004;31(7):1362-1368. PMID:15229958.
Namba K, Goto H, Kaburaki T, et al. A major review: current aspects of ocular Behçet’s disease in Japan. Ocul Immunol Inflamm. 2015;23(Suppl 1):S1-S23. doi:10.3109/09273948.2014.981547.
Pasadhika S, Rosenbaum JT.. Update on the use of systemic biologic agents in the treatment of noninfectious uveitis. Biologics. 2014;8:67-81. doi:10.2147/btt.s41477. PMID:24600203; PMCID:PMC3933243.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.