視神經炎

視神經脊髓炎譜系疾病(NMOSD)
一目瞭然的要點
Section titled “一目瞭然的要點”1. 什麼是視神經脊髓炎譜系疾病(NMOSD)?
Section titled “1. 什麼是視神經脊髓炎譜系疾病(NMOSD)?”視神經脊髓炎譜系疾病(NMOSD)是一種影響中樞神經系統的發炎性、抗體介導的自體免疫疾病。以前稱為「德維克病」,長期被視為多發性硬化症(MS)的亞型。然而,2004年發現針對水通道蛋白4(AQP4)的自體抗體(AQP4-IgG)後,它被確立為獨立的疾病實體。
歷史背景:1870年,Sir Thomas Clifford Allbutt首次描述脊髓炎與視神經病變的關聯。後續研究認識到這是一種不同於多發性硬化的疾病,現在被統稱為「NMOSD」。
流行病學如下:
- 發生率:AQP4陽性NMOSD的估計年發生率為每百萬人0.4~7.3例1)
- 性別差異:男女比例約為1:9,女性顯著居多
- 發病年齡:主要好發於中年(40~60歲)。在日本,高峰在30多歲後期至40多歲早期
- 種族:在非洲裔和亞洲裔人群中更常見1)
- 與懷孕的關聯:約20-47%的女性在懷孕期間或分娩/流產後一年內首次發病。
Q
NMOSD和多發性硬化症(MS)有何不同?
2. 主要症狀和臨床發現
Section titled “2. 主要症狀和臨床發現”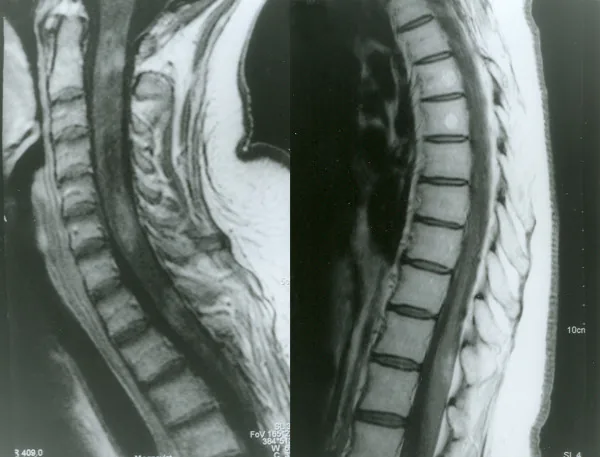
Petelin Gadze Z, et al. Patient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case report. BMC Neurol. 2009. Figure 2. PMCID: PMC2773232. License: CC BY.
2007年4月拍攝的脊髓釓增強T1加權MRI影像,顯示C2、C4-C5和Th1層級不均勻環狀增強,對應於本文「2. 主要症狀和臨床發現」部分討論的脊髓炎。
NMOSD的症狀因受影響部位而異。
- 急劇視力下降:主要症狀之一。特徵是對類固醇治療有抵抗性。
- 眼痛:伴隨視神經炎,約半數病例出現。
- 色覺異常:典型表現為紅色飽和度下降。
- 視野缺損:不僅限於中心暗點,還可能出現水平偏盲、雙顳側偏盲、同向偏盲。這是因為病變延伸至視交叉和視束。
- 感覺障礙和截癱:脊髓炎引起的運動和感覺障礙
- 膀胱直腸功能障礙:脊髓炎相關的自律神經功能障礙
- 頑固性呃逆和噁心嘔吐:最後區病變的特徵性症狀
- 眼球運動障礙:由腦幹病變引起
- 嗜睡症(猝睡症樣):由間腦/下視丘病變引起
發病初期可能出現類流感症狀(發燒、肌肉疼痛、頭痛)。
脊髓炎
最後區症候群
Q
NMOSD的視神經炎與MS相關視神經炎有何不同?
3. 原因與風險因素
Section titled “3. 原因與風險因素”NMOSD的確切病因尚未完全明瞭。一般認為其根本原因是自體免疫耐受性的喪失。
主要風險因素如下:
- 女性:男女比例約1:9,女性佔壓倒性多數
- 種族:亞洲人與非洲人發病風險較高
- 合併自體免疫疾病:全身性紅斑狼瘡(SLE)、修格連氏症候群、重症肌無力等
- 10–30%的NMOSD患者合併乾燥症候群。兒童病例也有報導5)
- 與重症肌無力的合併率為2–3%1)
- 惡性腫瘤(副腫瘤性NMOSD):估計3–5%的NMOSD為副腫瘤性2)3)
- 已報導的包括乳癌、肺癌和卵巢畸胎瘤等3)
- 有假說認為腫瘤內AQP4表現可誘發自體免疫反應2)
- 畸胎瘤相關NMOSD多見於年輕女性(平均年齡32.7歲)2)
4. 診斷與檢查方法
Section titled “4. 診斷與檢查方法”診斷標準(2015年國際共識)
Section titled “診斷標準(2015年國際共識)”AQP4-IgG陽性NMOSD的診斷標準需滿足以下三項:
- 至少一項核心臨床特徵
- AQP4-IgG陽性(使用最佳檢測方法)
- 排除其他診斷
AQP4-IgG陰性或未檢測之NMOSD診斷標準需滿足以下四項:
- 至少兩個核心臨床特徵(其中之一必須是視神經炎、LETM或最後區症候群)
- 空間多發性
- 滿足附加MRI要求
- 排除其他診斷
**主要臨床特徵(6項)**如下:
- 視神經炎
- 急性脊髓炎
- 最後區症候群(頑固性打嗝、噁心嘔吐)
- 急性腦幹症候群
- 症狀性猝睡症/急性間腦症候群
- 症狀性大腦症候群
下表顯示主要抗體檢測方法的比較。
| 檢測方法 | 敏感度 | 特異度 | 備註 |
|---|---|---|---|
| CBA(細胞基礎檢測) | 69.7~100% | 85.8~100% | 建議方法 |
| ELISA法 | 略遜於CBA | 略遜於CBA | 日本健保給付 |
- AQP4-IgG:NMOSD疾病特異性。建議在急性發作時及免疫抑制治療開始前檢測1)
- CBA(細胞基礎檢測):目前推薦的檢測方法。ELISA的偽陽性率是CBA的5倍1)
- MOG-IgG:約30%的AQP4-IgG陰性NMOSD患者呈陽性1)
- CSF寡克隆帶(OCB):NMOSD中為10–20%(MS中為88%)。陰性提示NMOSD1)
- CSF白血球計數:>50/μL、中性球或嗜酸性球的存在有助於區分NMOSD與MS
影像診斷(MRI)
Section titled “影像診斷(MRI)”- 脊髓MRI:LETM最具特徵性。中央灰質為主。伴有脊髓腫脹、T1低訊號和Gd顯影。約85%的AQP4+ NMOSD在急性脊髓炎時表現為LETM 1)
- 視神經MRI:必須使用脂肪抑制序列。雙側、長節段發炎(≥50%)為特徵。後部及視交叉受累是AQP4+ NMOSD的特徵 1)
- 腦部MRI:可見最後區病變、第四腦室周圍腦幹病變、下視丘/第三腦室周圍病變及廣泛白質病變
- NMOSD的特徵:與MS不同,無症狀的新發T2病變罕見(3–13%)。通常無需監測MRI 1)
- 多發性硬化症(MS)
- MOG抗體相關疾病(MOGAD)
- 急性瀰散性腦脊髓炎(ADEM)
- 全身性紅斑狼瘡
- 神經貝赫切特病
- 在老年人中,與缺血性視神經病變、頸椎病性脊髓病、脊髓梗塞和原發性中樞神經系統淋巴瘤的鑑別很重要
Q
即使AQP4抗體陰性,也能診斷為NMOSD嗎?
5. 標準治療方法
Section titled “5. 標準治療方法”首選:類固醇脈衝療法
- 甲基潑尼松龍1000mg/日靜脈滴注,連續3天
- 若視力改善不明顯,間隔3-4天後考慮再行一個療程。
- NMOSD的視神經炎對類固醇抗性高,若反應不充分,應及早考慮下一步治療。
二線治療:血漿置換療法
在類固醇脈衝治療無效時實施。以下方法為選項。
- 單純血漿置換(PE):效果最強,但對身體的傷害也最大。
- 雙重膜過濾血漿置換(DFPP)
- 免疫吸附療法(IA):可選擇性移除抗體。
效果順序為單純血漿交換 > 雙重膜濾過 > 免疫吸附。一個療程進行5~6次,治療後需住院管理直至體內IgG水平恢復。對於「視神經炎」,可能不在保險給付範圍內,需向患者說明。
復發預防(維持治療)
Section titled “復發預防(維持治療)”在AQP4+ NMOSD中,首次發作後應盡早開始維持治療1)。在日本,血漿交換後通常轉為prednisolone 510mg/日合併azathioprine 50100mg/日。
證據等級高的生物製劑如下:
補體抑制劑
- Eculizumab:900mg靜脈注射,每週1次×4次,然後每2週1次1200mg維持治療1)
- 拉武利珠單抗:根據體重的負荷劑量(2,400–3,000 mg)→ 從第15天起每8週給予3,000–3,600 mg1)
B細胞去除療法
- 利妥昔單抗:375mg/m² 靜脈注射,每週1次,共4次;或1000mg×2次(間隔2週)→ 之後每6個月給予1000mg×2次1)
- 伊奈利珠單抗:300mg 靜脈注射,每15天2次→每6個月一次1)
IL-6受體抑制劑
- 薩特利珠單抗:120mg 皮下注射,每4週一次1)
Q
如果類固醇脈衝療法無效怎麼辦?
A
血漿置換療法是下一個選擇。可選擇單純血漿置換、雙重膜過濾血漿置換或免疫吸附。單純血漿置換被認為效果最好,但對身體負擔也較大。一個療程進行5-6次,治療後需要住院管理。
6. 病理生理學與詳細致病機轉
Section titled “6. 病理生理學與詳細致病機轉”NMOSD本質上是一種星狀細胞病。致病機轉如下。
抗體產生與血腦屏障(BBB)通過
在外周,B細胞分化為分泌AQP4-IgG的漿母細胞。IL-6促進此分化並增加BBB通透性。最後區缺乏BBB,可能成為AQP4-IgG進入CNS的途徑。
星狀細胞損傷的連鎖反應
AQP4-IgG與星狀細胞足突上高度表現的AQP4水通道結合,透過以下途徑導致星狀細胞損傷。
- 補體經典途徑活化:AQP4-IgG的Fc部分激活補體,形成膜攻擊複合物(MAC),直接損傷星形膠質細胞。
- ADCC(抗體依賴性細胞毒性):NK細胞和中性粒細胞通過Fcγ受體損傷星形膠質細胞。
- C5a過敏毒素釋放:招募顆粒球(中性粒細胞、嗜酸性粒細胞),引起繼發性軸突損傷和脫髓鞘1)。
與MS的病理差異
MS以CD8+ T細胞為核心,以白質脫髓鞘為主;而NMOSD中CD4+ T細胞參與更多,形成累及灰質和白質的壞死性病變。
分佈原因
AQP4通道在視神經、最後區和脊髓中分布豐富,這些區域成為選擇性標靶。
生物標誌物
- 血清GFAP:反映星形膠細胞損傷,發作時升高
- 血清神經絲輕鏈(NfL):反映軸突損傷,與發作嚴重程度相關1)
涉及的細胞因子包括IL-6、IL-10、IL-17a、G-CSF、TNF-α和BAFF/APRIL。
7. 最新研究與未來展望(研究階段報告)
Section titled “7. 最新研究與未來展望(研究階段報告)”副腫瘤性NMOSD的機轉闡明與腫瘤篩檢
Section titled “副腫瘤性NMOSD的機轉闡明與腫瘤篩檢”估計3-5%的NMOSD為副腫瘤性。與卵巢畸胎瘤相關的病例尤其得到了詳細研究。
Ikeguchi等人(2021年)對6例卵巢畸胎瘤相關AQP4+ NMOSD病例進行了綜述2)。所有病例均為女性,平均發病年齡32.7歲(15-50歲)。6例中83%(5/6)出現噁心和嘔吐,83%腦脊髓液寡克隆區帶陽性,83%有背側腦幹病變。病理分析顯示,腫瘤內GFAP陽性神經組織中有AQP4免疫反應性和淋巴球浸潤,提示腫瘤內AQP4抗原呈遞觸發自體免疫反應的機轉。腫瘤切除後,60%(3/5)的病例AQP4-IgG轉為陰性。
Ding等人(2021)對43例副腫瘤性NMOSD進行了回顧3)。88.4%為女性,乳癌和肺癌是最常見的腫瘤類型。尤其強調50歲及以上NMOSD患者進行腫瘤篩查的重要性。
即使對於年輕患者,也建議進行包括畸胎瘤在內的腫瘤篩查。
難治性病例的免疫吸附療法
Section titled “難治性病例的免疫吸附療法”對於對類固醇、血漿置換和利妥昔單抗等治療無效的難治性NMOSD,已有報導顯示Protein-A免疫吸附療法(IA)的有效性。
Fan等人(2024)對一名35歲合併乾燥症候群且對類固醇脈衝和IVIG治療無效的難治性NMOSD女性患者實施了3次Protein-A免疫吸附療法4)。一週內,視力障礙、截癱和本體感覺障礙顯著改善,並確認AQP4-IgG、IgA、IgG和IgM迅速下降。4年追蹤期間未見復發或進展。
合併自體免疫疾病病例的特徵
Section titled “合併自體免疫疾病病例的特徵”NMOSD合併各種自體免疫疾病的實際情況正逐漸明朗。
Zhu等人(2025)報告了一例11歲發病的14歲女性NMOSD病例5)。該患者AQP4-IgG陽性,病程中確診合併原發性乾燥症。透過甲潑尼龍、IVIG、黴酚酸酯(MMF)轉換為他克莫司維持緩解。雖然成人NMOSD病例中20-30%合併自體免疫疾病,但該病例顯示兒童患者也可能出現合併症5)。
8. 參考文獻
Section titled “8. 參考文獻”- Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
- Ikeguchi R, Shimizu Y, Shimomura A, et al. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045.
- Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain Behav. 2021;11(9):e2282.
- Fan W, Chen X, Xiao P, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Front Immunol. 2024;15:1429405.
- Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.