El trastorno del espectro de la neuromielitis óptica (NMOSD) es una enfermedad inflamatoria, autoinmune y mediada por anticuerpos que afecta al sistema nervioso central. Anteriormente llamada “enfermedad de Devic”, durante mucho tiempo se consideró un subtipo de esclerosis múltiple (EM). Sin embargo, el descubrimiento de autoanticuerpos contra la acuaporina-4 (AQP4-IgG) en 2004 la estableció como una entidad patológica independiente.
Antecedentes históricos: En 1870, Sir Thomas Clifford Allbutt describió por primera vez la asociación entre mielitis y neuropatía óptica. Investigaciones posteriores lo reconocieron como una enfermedad distinta de la EM, y ahora se entiende bajo el concepto integral de “NMOSD”.
Epidemiología es la siguiente:
Incidencia: La incidencia anual estimada de NMOSD AQP4+ es de 0,4 a 7,3 por millón de personas1)
Sexo: La proporción hombre:mujer es de aproximadamente 1:9, con un marcado predominio femenino
Edad de inicio: Ocurre principalmente en la mediana edad (40–60 años). En Japón, el pico se da entre los 35 y 45 años
Raza: Tiende a ser más común en poblaciones africanas y asiáticas1)
Asociación con el embarazo: Aproximadamente el 20–47% de las mujeres experimentan su primer brote durante el embarazo o dentro del año posterior al parto o aborto espontáneo.
Q¿En qué se diferencia la NMOSD de la esclerosis múltiple (EM)?
A
La NMOSD es una enfermedad mediada por anticuerpos dirigidos contra el canal de agua AQP4 en los astrocitos, y su patología, tratamiento y pronóstico son fundamentalmente diferentes de la EM. En la NMOSD, la LETM y la neuritis óptica grave son típicas, y una diferencia importante es que los fármacos modificadores de la enfermedad efectivos para la EM, como el interferón beta, pueden desencadenar recaídas en la NMOSD.
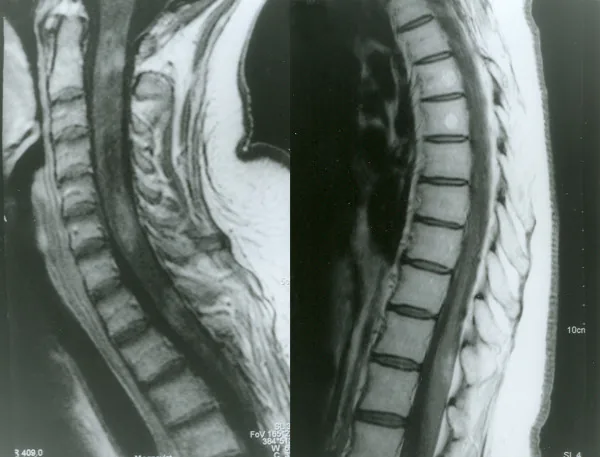
RM con contraste de la médula espinal en neuromielitis óptica
Petelin Gadze Z, et al. Patient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case report. BMC Neurol. 2009. Figure 2. PMCID: PMC2773232. License: CC BY.
Imagen de RM potenciada en T1 con gadolinio de la médula espinal tomada en abril de 2007, que muestra realce anular heterogéneo en los niveles C2, C4-C5 y Th1, correspondiente a la mielitis discutida en la sección “2. Síntomas principales y hallazgos clínicos”.
Los síntomas de la NMOSD varían según el área afectada.
Pérdida rápida de la visión: Uno de los síntomas principales. Se caracteriza por resistencia al tratamiento con esteroides.
Dolor ocular: Asociado a neuritis óptica, presente en aproximadamente la mitad de los casos.
Anomalía de la visión cromática: Típicamente una disminución de la saturación del rojo.
Defectos del campo visual: No se limitan a escotoma central; también pueden causar hemianopsia horizontal, hemianopsia bitemporal o hemianopsia homónima. Esto se debe a que las lesiones se extienden al quiasma óptico y al tracto óptico.
Alteración sensorial y paraplejía: Déficits motores y sensoriales debidos a mielitis
Disfunción vesical y rectal: Neuropatía autonómica asociada a mielitis
Hipo intratable y náuseas/vómitos: Síntomas característicos por lesión del área postrema
Alteración oculomotora: Debida a lesiones del tronco encefálico
Hipersomnia (similar a narcolepsia): Debida a lesiones diencefálicas/hipotalámicas
En la etapa inicial, pueden presentarse síntomas similares a la gripe (fiebre, mialgia, cefalea).
Afectación bilateral simultánea: La neuritis óptica en NMOSD es bilateral en el 17–82% de los casos. Una diferencia importante con la EM.
Pérdida visual grave: En NMOSD con AQP4+, la mediana de la agudeza visual mínima es de movimiento de manos (HM). Tras la recuperación, la mediana sigue siendo cuenta dedos, y el 60–69% presenta discapacidad visual permanente de 20/200 o peor en al menos un ojo.
Mielitis
LETM (Mielitis Transversa Longitudinalmente Extensa): Lesión continua que abarca 3 o más segmentos vertebrales. Aproximadamente el 85% de los casos de NMOSD con AQP4+ lo presentan durante la mielitis aguda.
Síndrome medular completo: Afecta las tres vías: motora, sensitiva y autónoma.
Deterioro funcional grave: Más del 30% de los pacientes dependen de silla de ruedas en el punto más bajo del ataque. El 37–44% de los pacientes con NMOSD AQP4+ finalmente requieren ayudas para caminar.
Síndrome del área postrema
Hipo intratable: Persiste durante días a semanas y no responde a los antieméticos habituales.
Náuseas y vómitos: Esto se debe a que el área postrema carece de barrera hematoencefálica, lo que facilita que AQP4-IgG llegue directamente a esta zona.
Hallazgos diagnósticos clave de NMOSD: El hipo intratable inexplicable es un motivo para sospechar activamente NMOSD.
Q¿En qué se diferencia la neuritis óptica en NMOSD de la relacionada con EM?
A
La neuritis óptica en NMOSD es más grave, bilateral y recurrente, con mal pronóstico visual. En NMOSD AQP4+, se informa que el 60–69% de los pacientes presentan discapacidad visual permanente de 20/200 o peor en al menos un ojo. Además, tiende a afectar el quiasma óptico, causando diversos defectos del campo visual como hemianopsia bitemporal, lo que también la diferencia de la EM.
RM de médula espinal: La LETM es la más característica. Predominio de la sustancia gris central. Acompañada de hinchazón de la médula espinal, hipointensidad en T1 y realce con Gd. Aproximadamente el 85% de los NMOSD AQP4+ presentan LETM durante la mielitis aguda 1)
RM de nervio óptico: La supresión grasa es esencial. La inflamación bilateral y de segmento largo (≥50%) es característica. La afectación de la porción posterior y el quiasma óptico es característica del NMOSD AQP4+ 1)
RM cerebral: Se observan lesiones del área postrema, lesiones del tronco encefálico periventriculares del cuarto ventrículo, lesiones hipotalámicas/periventriculares del tercer ventrículo y lesiones extensas de la sustancia blanca
Características del NMOSD: A diferencia de la EM, las nuevas lesiones asintomáticas en T2 son raras (3–13%). Por lo general, no se necesita RM de vigilancia 1)
En pacientes de edad avanzada, es importante diferenciar de neuropatía óptica isquémica, mielopatía espondilótica cervical, infarto de médula espinal y linfoma primario del SNC
Q¿Se puede diagnosticar NMOSD incluso si el anticuerpo AQP4 es negativo?
A
Incluso si AQP4-IgG es negativo o no se ha analizado, se puede diagnosticar NMOSD si se cumplen dos o más características clínicas principales, se satisfacen los requisitos adicionales de resonancia magnética y se excluyen otras enfermedades. Además, aproximadamente el 30% de los casos negativos para AQP4-IgG son positivos para MOG-IgG, por lo que se recomienda medir ambos anticuerpos. Menos del 1% de los casos negativos para AQP4-IgG seroconvierten posteriormente.
Metilprednisolona 1,000 mg/día por vía intravenosa durante 3 días
Si no hay mejoría visual, considerar repetir un ciclo después de un intervalo de 3 a 4 días.
La neuritis óptica en NMOSD es altamente resistente a los esteroides, por lo que si la respuesta es insuficiente, considere el siguiente tratamiento temprano.
Segunda línea: Terapia de intercambio plasmático
Se realiza cuando no hay respuesta a la pulsoterapia con esteroides. Las siguientes son opciones.
Intercambio plasmático simple (PE): Más efectivo pero también causa el mayor daño al cuerpo.
Filtración plasmática de doble membrana (DFPP)
Terapia de inmunoadsorción (IA): Permite la eliminación selectiva de anticuerpos.
El orden de efectividad es: recambio plasmático simple > filtración de doble membrana > inmunoadsorción. Un ciclo consta de 5 a 6 sesiones, y después del tratamiento se requiere hospitalización hasta que los niveles de IgG en el cuerpo se recuperen. Tenga en cuenta que para la “neuritis óptica” puede no estar cubierto por el seguro, por lo que es necesario explicarlo al paciente.
En NMOSD con AQP4+, se debe iniciar la terapia de mantenimiento temprano después del primer ataque 1). En Japón, es común cambiar a prednisolona 5–10 mg/día más azatioprina 50–100 mg/día después del recambio plasmático.
Los fármacos biológicos con alto nivel de evidencia son los siguientes:
Inhibidores del complemento
Eculizumab: 900 mg IV semanal × 4 dosis, luego 1,200 mg cada 2 semanas como mantenimiento 1)
Ravulizumab: Dosis de carga basada en el peso (2,400–3,000 mg) → 3,000–3,600 mg cada 8 semanas a partir del día 151)
Terapia de depleción de células B
Rituximab: 375 mg/m² IV semanal × 4 dosis, o 1,000 mg × 2 dosis (con 2 semanas de diferencia) → luego 1,000 mg × 2 dosis cada 6 meses1)
Inebilizumab: 300 mg IV dos veces cada 15 días → cada 6 meses1)
Inhibidor del receptor de IL-6
Satralizumab: 120 mg inyección subcutánea cada 4 semanas1)
Q¿Qué hacer si la terapia con pulsos de esteroides no es efectiva?
A
La plasmaféresis es la siguiente opción. Se elige entre plasmaféresis simple, plasmaféresis de doble filtración o inmunoadsorción. La plasmaféresis simple se considera la más efectiva, pero también supone una mayor carga para el organismo. Se realizan de 5 a 6 sesiones por ciclo, y se requiere hospitalización después del tratamiento.
La NMOSD es esencialmente una astrocitopatía. La patogenia es la siguiente.
Producción de anticuerpos y paso a través de la barrera hematoencefálica (BHE)
En la periferia, los linfocitos B se diferencian en plasmablastos secretores de AQP4-IgG. La IL-6 promueve esta diferenciación y aumenta la permeabilidad de la BHE. El área postrema carece de BHE y puede ser una vía de entrada de AQP4-IgG al SNC.
Cascada de daño astrocitario
La AQP4-IgG se une a los canales de agua AQP4 altamente expresados en los pies de los astrocitos, lo que provoca daño astrocitario a través de las siguientes vías.
Activación de la vía clásica del complemento: La porción Fc de AQP4-IgG activa el complemento, formando el complejo de ataque a la membrana (MAC) y dañando directamente los astrocitos.
ADCC (citotoxicidad celular dependiente de anticuerpos): Las células NK y los neutrófilos dañan los astrocitos a través de los receptores Fcγ.
Liberación de anafilatoxina C5a: Recluta granulocitos (neutrófilos, eosinófilos), causando daño axonal secundario y desmielinización1).
Diferencias patológicas con la EM
En la EM, los linfocitos T CD8+ desempeñan un papel central con desmielinización predominante de la sustancia blanca, mientras que en la NMOSD, los linfocitos T CD4+ están más involucrados, formando lesiones necróticas que afectan tanto a la sustancia gris como a la blanca.
Razón de la distribución
Los canales AQP4 se distribuyen abundantemente en el nervio óptico, el área postrema y la médula espinal, lo que hace que estas regiones sean selectivamente atacadas.
Biomarcadores
GFAP sérica: Refleja daño astrocitario y se eleva durante los ataques
Cadena ligera de neurofilamentos sérica (NfL): Refleja daño axonal y se correlaciona con la gravedad del ataque1)
Las citocinas implicadas incluyen IL-6, IL-10, IL-17a, G-CSF, TNF-α y BAFF/APRIL.
7. Investigación más reciente y perspectivas futuras (Informes en fase de investigación)
Se estima que el 3-5% de los casos de NMOSD son paraneoplásicos. Los casos asociados con teratoma ovárico se han estudiado con especial detalle.
Ikeguchi et al. (2021) realizaron una revisión de 6 casos de NMOSD AQP4+ asociada a teratoma ovárico2). Todos los casos eran mujeres, con una edad media de inicio de 32.7 años (rango 15-50 años). De los 6 casos, el 83% (5/6) presentó náuseas y vómitos, el 83% tuvo bandas oligoclonales positivas en LCR y el 83% presentó lesiones en el tronco encefálico dorsal. El análisis patológico reveló inmunorreactividad para AQP4 e infiltración linfocítica en tejido neural positivo para GFAP dentro del tumor, lo que sugiere un mecanismo en el que la presentación de antígeno AQP4 dentro del tumor desencadena una respuesta autoinmune. Tras la resección tumoral, la AQP4-IgG se negativizó en el 60% (3/5) de los casos.
Ding et al. (2021) realizaron una revisión de 43 casos de NMOSD paraneoplásica 3). El 88.4% eran mujeres, y el cáncer de mama y de pulmón fueron los tipos tumorales más frecuentes. Se enfatiza la importancia del cribado tumoral, especialmente en pacientes con NMOSD de 50 años o más.
Se recomienda el cribado de tumores, incluidos los teratomas, incluso en pacientes jóvenes.
Terapia de inmunoadsorción para casos refractarios
Se ha informado la eficacia de la terapia de inmunoadsorción con proteína A (IA) para la NMOSD refractaria que no responde a esteroides, plasmaféresis y rituximab.
Fan et al. (2024) realizaron tres sesiones de terapia de inmunoadsorción con proteína A en una mujer de 35 años con NMOSD refractaria asociada a síndrome de Sjögren que no respondió a pulsos de esteroides ni a IVIG4). En una semana, la discapacidad visual, la paraplejía y los déficits de sensibilidad propioceptiva mejoraron notablemente, y se confirmó una rápida disminución de AQP4-IgG, IgA, IgG e IgM. No se observó recurrencia ni progresión durante 4 años de seguimiento.
Características de los casos con enfermedades autoinmunes comórbidas
La realidad de la coexistencia de diversas enfermedades autoinmunes con NMOSD se está aclarando.
Zhu et al. (2025) reportaron el caso de una niña de 14 años que desarrolló NMOSD a los 11 años5). Era AQP4-IgG positiva y se confirmó la coexistencia de síndrome de Sjögren primario durante el curso. Se mantuvo la remisión cambiando de metilprednisolona, IVIG y micofenolato de mofetilo (MMF) a tacrolimus. Aunque se ha informado que el 20-30% de los casos de NMOSD en adultos presentan enfermedades autoinmunes concomitantes, este caso demuestra que también ocurren en pacientes pediátricos5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, et al. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain Behav. 2021;11(9):e2282.
Fan W, Chen X, Xiao P, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Front Immunol. 2024;15:1429405.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.