Оптиконевромиелит спектра расстройств (NMOSD) — это воспалительное, антител-опосредованное аутоиммунное заболевание, поражающее центральную нервную систему. Ранее называлось «болезнью Девика» и долгое время считалось подтипом рассеянного склероза (РС). Однако в 2004 году с открытием аутоантител к аквапорину-4 (AQP4-IgG) оно было признано самостоятельной нозологической единицей.
Историческая справка: в 1870 году сэр Томас Клиффорд Оллбатт впервые описал связь между миелитом и поражением зрительного нерва. Последующие исследования показали, что это заболевание отличается от РС, и в настоящее время оно рассматривается в рамках общего понятия «NMOSD».
Эпидемиология следующая:
Заболеваемость: предполагаемая годовая заболеваемость AQP4+NMOSD составляет 0,4–7,3 на миллион человек1)
Пол: соотношение мужчин и женщин составляет примерно 1:9 с выраженным преобладанием женщин
Возраст начала: преимущественно средний возраст (40–60 лет). В Японии пик приходится на возраст от 35 до 45 лет
Раса: чаще встречается у лиц африканского и азиатского происхождения1)
Связь с беременностью: примерно у 20–47% женщин заболевание впервые проявляется во время беременности или в течение года после родов или выкидыша.
QЧем отличается NMOSD от рассеянного склероза (РС)?
A
NMOSD — это антитело-опосредованное заболевание, направленное против аквапориновых водных каналов AQP4 на астроцитах, и его патогенез, лечение и прогноз принципиально отличаются от РС. Для NMOSD типичны LETM и тяжелый оптический неврит, а также важным отличием является то, что препараты, изменяющие течение РС, такие как интерферон-β, эффективные при РС, могут провоцировать рецидивы NMOSD.
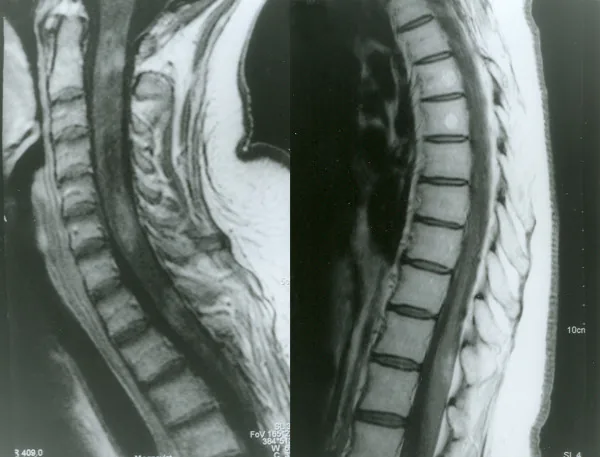
МРТ спинного мозга с контрастированием при оптикомиелите
Petelin Gadze Z, et al. Patient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case report. BMC Neurol. 2009. Figure 2. PMCID: PMC2773232. License: CC BY.
Гадолиний-усиленная T1-взвешенная МРТ спинного мозга, выполненная в апреле 2007 года, демонстрирующая неравномерное кольцевидное усиление на уровнях C2, C4-C5 и Th1. Это соответствует миелиту, описанному в разделе «2. Основные симптомы и клинические проявления».
Симптомы NMOSD разнообразны и зависят от пораженной области.
Резкое снижение зрения: один из основных симптомов. Характерна устойчивость к лечению стероидами
Боль в глазу: сопровождает неврит зрительного нерва, наблюдается примерно в половине случаев
Нарушение цветоощущения: типично снижение насыщенности красного цвета
Дефекты поля зрения: не ограничиваются центральной скотомой, могут возникать горизонтальная гемианопсия, битемпоральная гемианопсия, гомонимная гемианопсия. Это связано с распространением поражения на хиазму и зрительный тракт
Нарушения чувствительности, парапарез: двигательные и чувствительные нарушения вследствие миелита
Нарушения функции мочевого пузыря и прямой кишки: вегетативные нарушения, связанные с миелитом
Рефрактерная икота, тошнота и рвота: характерные симптомы при поражении area postrema
Нарушения движений глаз: вследствие поражения ствола мозга
Гиперсомния (нарколепсия): вследствие поражения гипоталамуса/диэнцефальной области
В начале заболевания могут наблюдаться гриппоподобные симптомы (лихорадка, миалгия, головная боль).
Отек диска зрительного нерва: наблюдается в острой фазе, впоследствии переходит в атрофию зрительного нерва.
RAPD (относительный афферентный зрачковый дефект): выявляется одно- или двусторонне.
Двустороннее одновременное поражение: при NMOSD неврит зрительного нерва в 17–82% случаев является двусторонним. Важное отличие от РС.
Тяжелое снижение зрения: при AQP4+NMOSD медиана минимальной остроты зрения соответствует счету пальцев у лица (HM). После восстановления медиана составляет счет пальцев, у 60–69% пациентов сохраняется стойкое снижение зрения до 20/200 или хуже хотя бы на одном глазу.
Миелит
Продольный поперечный миелит (LETM): непрерывное поражение, охватывающее 3 и более позвонков. Около 85% случаев AQP4+NMOSD проявляются этим при остром миелите.
Полный спинальный синдром: вовлечение всех трех путей — двигательного, чувствительного и вегетативного.
Тяжелые функциональные нарушения: более 30% пациентов на пике приступа становятся зависимыми от инвалидной коляски. 37–44% пациентов с AQP4+NMOSD в конечном итоге нуждаются в средствах для ходьбы.
Синдром area postrema
Рефрактерная икота: длится от нескольких дней до недель, не поддается действию обычных противорвотных средств.
Тошнота и рвота: обусловлены тем, что area postrema лишена гематоэнцефалического барьера, что делает ее легкодоступной для AQP4-IgG.
Ключевой диагностический признак NMOSD: необъяснимая рефрактерная икота является поводом активно подозревать NMOSD.
QЧем отличается оптический неврит при NMOSD от такового при РС?
A
Оптический неврит при NMOSD более тяжелый, двусторонний, рецидивирующий и имеет худший прогноз для зрения. При AQP4+ NMOSD у 60–69% пациентов сохраняется стойкое снижение зрения до 20/200 или хуже хотя бы на один глаз. Также часто вовлекается хиазма, что приводит к разнообразным дефектам поля зрения, таким как битемпоральная гемианопсия, что отличает его от РС.
МРТ спинного мозга: наиболее характерен LETM. Преимущественное поражение центрального серого вещества. Сопровождается отеком спинного мозга, гипоинтенсивностью на T1-ВИ и Gd-усилением. Примерно у 85% пациентов с AQP4+NMOSD наблюдается LETM при остром миелите1)
МРТ зрительного нерва: обязательно использование подавления жира. Характерно двустороннее воспаление на большом протяжении (>50%). Вовлечение задних отделов и хиазмы характерно для AQP4+NMOSD1)
МРТ головного мозга: выявляются поражения area postrema, ствола мозга вокруг четвертого желудочка, гипоталамуса/области вокруг третьего желудочка, обширные поражения белого вещества и др.
Особенности NMOSD: в отличие от РС, бессимптомные новые очаги на Т2-ВИ редки (3–13%). Обычно контрольная МРТ не требуется1)
У пожилых пациентов важно дифференцировать с ишемической оптической нейропатией, шейной миелопатией, спинальным инфарктом и первичной лимфомой ЦНС.
QМожет ли быть диагностировано NMOSD при отрицательных антителах к AQP4?
A
Да. Даже при отрицательных AQP4-IgG или отсутствии тестирования, если выполнены два или более основных клинических признаков, дополнительные критерии МРТ и исключены другие заболевания, может быть диагностировано NMOSD. Кроме того, примерно у 30% AQP4-IgG-отрицательных пациентов выявляются MOG-IgG, поэтому рекомендуется определение обоих антител. Менее 1% AQP4-IgG-отрицательных пациентов впоследствии сероконвертируют.
Внутривенное введение метилпреднизолона в дозе 1000 мг/сут в течение 3 дней
При отсутствии улучшения зрения рассмотреть повторный курс через 3–4 дня
Неврит зрительного нерва при NMOSD часто устойчив к стероидам, поэтому при недостаточном ответе следует рассмотреть следующее лечение на ранней стадии.
Вторая линия: плазмаферез
Проводится при отсутствии реакции на пульс-терапию стероидами. Доступны следующие методы.
Простой плазмаферез (ПЭ): наиболее эффективен, но также наносит наибольший ущерб организму.
Двойная фильтрация плазмы (ДФПП)
Иммуноадсорбция (ИА): позволяет селективно удалять антитела.
Эффективность убывает в порядке: простой плазмаферез > двойная фильтрация > иммуноадсорбция. Проводится 5–6 сеансов за курс, после лечения необходима госпитализация до восстановления уровня IgG в организме. Следует отметить, что при «неврите зрительного нерва» эти методы могут не покрываться страховкой, что требует разъяснения пациенту.
При AQP4+NMOSD рекомендуется начинать поддерживающую терапию рано после первого приступа 1). В Японии после плазмафереза обычно переходят на преднизолон 5–10 мг/сут + азатиоприн 50–100 мг/сут.
Биологические препараты с высоким уровнем доказательности включают:
Ингибиторы комплемента
Экулизумаб: 900 мг в/в еженедельно × 4, затем 1200 мг каждые 2 недели в качестве поддерживающей терапии 1)
Равулизумаб: нагрузочная доза на основе массы тела (2400–3000 мг), затем с 15-го дня 3000–3600 мг каждые 8 недель 1)
Терапия, направленная на удаление B-клеток
Ритуксимаб: 375 мг/м² в/в еженедельно × 4, или 1000 мг × 2 (с интервалом 2 недели), затем 1000 мг × 2 каждые 6 месяцев 1)
Инебилизумаб: 300 мг в/в каждые 15 дней × 2, затем каждые 6 месяцев 1)
Ингибитор рецептора интерлейкина-6
Сатрализумаб: 120 мг подкожно каждые 4 недели1)
QЧто делать, если пульс-терапия стероидами неэффективна?
A
Следующим вариантом является плазмаферез. Выбирается один из трех типов: простой плазмаферез, двойная фильтрационная плазмаферез или иммуноадсорбция. Простой плазмаферез считается наиболее эффективным, но также оказывает наибольшую нагрузку на организм. Проводится 5–6 сеансов за курс, после лечения требуется госпитализация.
NMOSD по своей сути является астроцитопатией. Механизм развития следующий.
Продукция антител и прохождение через гематоэнцефалический барьер (ГЭБ)
На периферии B-клетки дифференцируются в плазмабласты, секретирующие AQP4-IgG. IL-6 способствует этой дифференцировке и повышает проницаемость ГЭБ. Область postrema лишена ГЭБ и может служить путем проникновения AQP4-IgG в ЦНС.
Каскад повреждения астроцитов
AQP4-IgG связывается с водными каналами AQP4, которые в высокой плотности экспрессируются на отростках астроцитов, что приводит к повреждению астроцитов по следующим путям.
Активация классического пути комплемента: Fc-часть AQP4-IgG активирует комплемент, образуя мембраноатакующий комплекс (MAC), который напрямую повреждает астроциты
ADCC (антителозависимая клеточная цитотоксичность): NK-клетки и нейтрофилы повреждают астроциты через Fcγ-рецепторы
Высвобождение анафилатоксина C5a: привлекает гранулоциты (нейтрофилы и эозинофилы), вызывая вторичное повреждение аксонов и демиелинизацию1)
Патологические различия с РС
При РС преобладает демиелинизация белого вещества с центральной ролью CD8+ Т-клеток, тогда как при NMOSD большую роль играют CD4+ Т-клетки, формирующие некротические поражения как серого, так и белого вещества.
Причина распределения
Каналы AQP4 в большом количестве расположены в зрительном нерве, area postrema и спинном мозге, поэтому эти области избирательно поражаются.
Биомаркеры
Сывороточный GFAP: отражает повреждение астроцитов, повышается во время приступа
Сывороточная легкая цепь нейрофиламентов (NfL): отражает повреждение аксонов, коррелирует с тяжестью приступа1)
Сообщается, что участвующими цитокинами являются IL-6, IL-10, IL-17a, G-CSF, TNF-α и BAFF/APRIL.
7. Новейшие исследования и перспективы на будущее (отчеты на стадии исследований)
По оценкам, 3–5% случаев NMOSD являются паранеопластическими. Особенно подробно изучены случаи, связанные с тератомой яичника.
Ikeguchi и соавт. (2021) провели обзор 6 случаев AQP4+NMOSD, ассоциированных с тератомой яичника2). Все пациенты — женщины, средний возраст начала заболевания 32,7 года (от 15 до 50 лет). У 83% (5/6) пациентов наблюдались тошнота и рвота, у 83% были положительные олигоклональные полосы в ЦСЖ, у 83% выявлены поражения дорсального отдела ствола мозга. Патологический анализ показал наличие AQP4-иммунореактивности и лимфоцитарной инфильтрации в GFAP-положительной нервной ткани внутри опухоли, что предполагает механизм, при котором презентация антигена AQP4 в опухоли запускает аутоиммунную реакцию. После удаления опухоли у 60% (3/5) пациентов произошла сероконверсия AQP4-IgG.
Ding и соавт. (2021) провели обзор 43 случаев паранеопластической NMOSD3). 88,4% составили женщины, наиболее частыми типами опухолей были рак молочной железы и легких. Особо подчеркивается важность скрининга на опухоли у пациентов с NMOSD старше 50 лет.
Даже у молодых пациентов рекомендуется скрининг на опухоли, включая тератомы.
Иммуноадсорбционная терапия при рефрактерных случаях
Сообщается об эффективности иммуноадсорбционной терапии с белком A (IA) при рефрактерной NMOSD, не отвечающей на стероиды, плазмаферез и ритуксимаб.
Fan и соавт. (2024) провели 3 сеанса иммуноадсорбции с белком A у 35-летней женщины с рефрактерной NMOSD в сочетании с синдромом Шегрена, не отвечавшей на пульс-терапию стероидами и ВВИГ 4). В течение недели отмечено значительное улучшение нарушений зрения, парапареза и проприоцептивных расстройств, а также быстрое снижение уровней AQP4-IgG, IgA, IgG и IgM. За 4 года наблюдения рецидивов и прогрессирования не выявлено.
Особенности случаев с сочетанием аутоиммунных заболеваний
Постепенно выясняется реальная картина сочетания NMOSD с различными аутоиммунными заболеваниями.
Zhu и соавт. (2025) сообщили о случае 14-летней девочки, у которой NMOSD развилась в возрасте 11 лет 5). Был выявлен положительный AQP4-IgG, а в ходе течения подтвердилось сочетание с первичным синдромом Шегрена. Ремиссия поддерживается с помощью метилпреднизолона, ВВИГ и перехода с микофенолата мофетила (ММФ) на такролимус. Хотя сообщается о сочетании с аутоиммунными заболеваниями у 20–30% взрослых пациентов с NMOSD, данный случай показывает, что такое сочетание возможно и у детей 5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, et al. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain Behav. 2021;11(9):e2282.
Fan W, Chen X, Xiao P, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Front Immunol. 2024;15:1429405.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.