اختلال طیف نورومیلیت اپتیکا (NMOSD) یک بیماری التهابی، آنتیبادیمحور و خودایمنی است که سیستم عصبی مرکزی را درگیر میکند. پیشتر به عنوان «بیماری دویک» شناخته میشد و سالها زیرگروهی از مولتیپل اسکلروزیس (MS) محسوب میشد. اما در سال ۲۰۰۴ با کشف آنتیبادی خودی علیه آکواپورین ۴ (AQP4-IgG) به عنوان یک واحد بیماری مستقل تثبیت شد.
پیشینه تاریخی: در سال ۱۸۷۰، سر توماس کلیفورد آلبوت برای اولین بار ارتباط بین میلیت و اختلال بینایی را توصیف کرد. پژوهشهای بعدی آن را به عنوان بیماری متفاوت از MS شناسایی کرد و امروزه با مفهوم جامع «NMOSD» در نظر گرفته میشود.
همهگیریشناسی به شرح زیر است:
بروز: بروز سالانه تخمینی NMOSD مثبت AQP4 بین ۰.۴ تا ۷.۳ در هر میلیون نفر 1)
نسبت جنسی: نسبت زن به مرد حدود ۱:۹ است و زنان به طور قابل توجهی بیشتر مبتلا میشوند
سن شروع: عمدتاً در میانسالی (۴۰ تا ۶۰ سال) شایع است. در ژاپن، اوج بروز در اواخر دهه ۳۰ تا اوایل دهه ۴۰ زندگی است
نژاد: تمایل بیشتر در افراد آفریقاییتبار و آسیاییتبار 1)
ارتباط با بارداری: حدود ۲۰ تا ۴۷ درصد از زنان اولین حمله خود را در دوران بارداری یا در عرض یک سال پس از زایمان یا سقط تجربه میکنند.
Qتفاوت NMOSD و مولتیپل اسکلروزیس (MS) چیست؟
A
NMOSD یک بیماری با واسطه آنتیبادی است که کانال آبی AQP4 در آستروسیتها را هدف قرار میدهد و از نظر پاتوفیزیولوژی، درمان و پیشآگهی با MS تفاوت اساسی دارد. در NMOSD، LETM و نوریت اپتیک شدید معمول است و داروهای تعدیلکننده بیماری مانند اینترفرون بتا که در MS مؤثر هستند، ممکن است باعث عود NMOSD شوند که این نیز یک تفاوت مهم است.
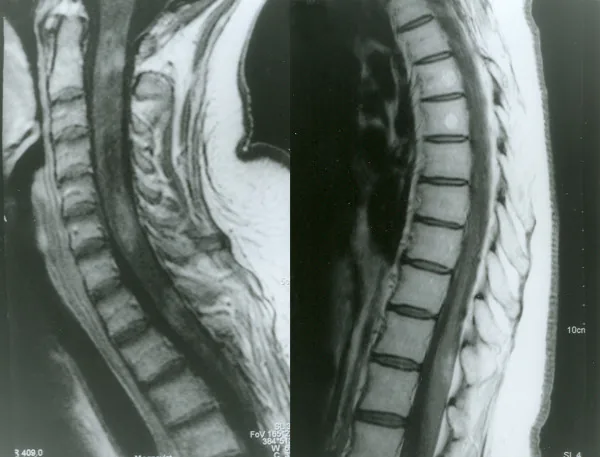
Petelin Gadze Z, et al. Patient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case report. BMC Neurol. 2009. Figure 2. PMCID: PMC2773232. License: CC BY.
تصویر MRI T1-weighted با گادولینیوم از نخاع که در آوریل ۲۰۰۷ گرفته شده است و افزایش کنتراست حلقوی ناهمگن در سطوح C2، C4-C5 و Th1 را نشان میدهد. این تصویر مربوط به میلیت است که در بخش «۲. علائم اصلی و یافتههای بالینی» بحث شده است.
ادم پاپی: در مرحله حاد دیده میشود و بعداً به آتروفی عصب بینایی تبدیل میشود.
RAPD (نقص نسبی پاسخ مردمک): به صورت یکطرفه یا دوطرفه دیده میشود.
دو طرفه همزمان: نوریت اپتیک در NMOSD در ۱۷ تا ۸۲٪ موارد دو طرفه همزمان است. تفاوت مهم با MS.
کاهش شدید بینایی: در AQP4+NMOSD، میانه حدت بینایی در بدترین حالت در حد حرکت دست (HM) است. حتی پس از بهبودی، میانه حدت بینایی در حد شمارش انگشتان باقی میماند و ۶۰ تا ۶۹٪ موارد حداقل در یک چشم دچار اختلال بینایی دائمی ۲۰/۲۰۰ یا بدتر میشوند.
میلیت (التهاب نخاع)
LETM (میلیت عرضی طولانی): ضایعه پیوسته به طول سه یا بیش از سه مهره. حدود ۸۵٪ موارد AQP4+NMOSD در زمان میلیت حاد این الگو را نشان میدهند.
سندرم کامل نخاعی: درگیری هر سه مسیر حرکتی، حسی و خودمختار.
ناتوانی شدید: بیش از ۳۰٪ بیماران در بدترین نقطه حمله به ویلچر وابسته میشوند. ۳۷ تا ۴۴٪ موارد AQP4+NMOSD در نهایت به وسایل کمک حرکتی نیاز پیدا میکنند.
سندرم ناحیه پوسترما (Area Postrema)
سکسکه مقاوم: چند روز تا چند هفته ادامه یافته و به داروهای معمول ضد تهوع پاسخ نمیدهد.
تهوع و استفراغ: به دلیل فقدان سد خونی-مغزی در ناحیه پوسترما، AQP4-IgG به راحتی به این ناحیه دسترسی پیدا میکند.
یافته کلیدی تشخیصی NMOSD: سکسکه مقاوم بدون علت مشخص، یک نشانه قوی برای شک به NMOSD است.
Qنوریت اپتیک در NMOSD چه تفاوتی با نوع مرتبط با MS دارد؟
A
نوریت اپتیک در NMOSD شدیدتر، دوطرفه و عودکننده است و پیشآگهی بینایی ضعیفتری دارد. در AQP4+ NMOSD، ۶۰ تا ۶۹٪ بیماران حداقل در یک چشم دچار کاهش دائمی بینایی تا ۲۰/۲۰۰ یا بدتر میشوند. همچنین درگیری کیاسمای بینایی شایعتر است و باعث نقایص متنوع میدان بینایی مانند همیانوپسی دوتمپورال میشود که از تفاوتهای آن با MS است.
MRI نخاع: LETM مشخصترین ویژگی است. غالباً در ماده خاکستری مرکزی. همراه با تورم نخاع، سیگنال پایین T1 و اثر کنتراست Gd. حدود 85% از AQP4+NMOSD در هنگام میلیت حاد LETM نشان میدهند1)
MRI عصب بینایی: تصاویر با سرکوب چربی ضروری است. التهاب دوطرفه و طولانی (بیش از 50%) مشخصه است. درگیری بخش خلفی و کیاسمای بینایی برای AQP4+NMOSD مشخص است1)
در افراد مسن، افتراق از نوروپاتی ایسکمیک بینایی، میلوپاتی اسپوندیلوتیک گردنی، انفارکتوس نخاعی و لنفوم اولیه CNS اهمیت دارد.
Qآیا حتی اگر آنتیبادی AQP4 منفی باشد، NMOSD تشخیص داده میشود؟
A
بله. حتی در موارد AQP4-IgG منفی یا آزمایش نشده، در صورت داشتن دو یا بیشتر از ویژگیهای بالینی اصلی، برآورده کردن معیارهای اضافی MRI و رد سایر بیماریها، NMOSD تشخیص داده میشود. همچنین، حدود 30% از موارد AQP4-IgG منفی، MOG-IgG مثبت هستند و اندازهگیری هر دو آنتیبادی توصیه میشود. کمتر از 1% از موارد AQP4-IgG منفی بعداً سرکونورت میشوند.
ترتیب اثربخشی به صورت پلاسمافرزیس ساده > فیلتراسیون دو غشایی > ایمونوادسورپشن است. یک دوره ۵ تا ۶ جلسه انجام میشود و پس از درمان تا زمان بازگشت سطح IgG بدن نیاز به بستری است. توجه داشته باشید که برای «نوریت اپتیک» ممکن است تحت پوشش بیمه نباشد و نیاز به توضیح به بیمار است.
در AQP4+NMOSD، توصیه میشود درمان نگهدارنده بلافاصله پس از اولین حمله شروع شود1). در ژاپن، پس از تعویض پلاسما، انتقال به پردنیزولون ۵-۱۰ میلیگرم/روز + آزاتیوپرین ۵۰-۱۰۰ میلیگرم/روز رایج است.
داروهای بیولوژیک با سطح شواهد بالا به شرح زیر هستند:
مهارکنندههای کمپلمان
اکولیزوماب: ۹۰۰ میلیگرم وریدی هفتهای یک بار × ۴ نوبت، سپس ۱۲۰۰ میلیگرم هر دو هفته یک بار به عنوان دوز نگهدارنده1)
راولیزوماب: دوز بارگیری بر اساس وزن (۲۴۰۰-۳۰۰۰ میلیگرم)، سپس از روز ۱۵ به بعد ۳۰۰۰-۳۶۰۰ میلیگرم هر ۸ هفته1)
درمان حذف سلولهای B
ریتوکسیماب: ۳۷۵ میلیگرم/متر مربع وریدی هفتهای یک بار × ۴ نوبت، یا ۱۰۰۰ میلیگرم × ۲ نوبت (با فاصله ۲ هفته)، سپس هر ۶ ماه ۱۰۰۰ میلیگرم × ۲ نوبت1)
اینبلیزوماب: ۳۰۰ میلیگرم وریدی هر ۱۵ روز دو بار، سپس هر ۶ ماه یک بار1)
مهارکنندههای گیرنده IL-6
ساترالیزوماب: 120 میلیگرم تزریق زیرجلدی، هر 4 هفته یک بار1)
Qاگر درمان با پالس استروئید مؤثر نباشد چه باید کرد؟
A
پلاسمافرز گزینه بعدی است. از بین سه نوع پلاسمافرز ساده، پلاسمافرز با فیلتر غشایی دوگانه و جذب ایمنی انتخاب میشود. پلاسمافرز ساده مؤثرترین در نظر گرفته میشود اما بار بیشتری بر بدن دارد. یک دوره شامل 5 تا 6 جلسه است و پس از درمان نیاز به بستری در بیمارستان است.
NMOSD اساساً یک بیماری آستروسیتی (آستروسیتوپاتی) است. مکانیسم بیماری به شرح زیر است.
تولید آنتیبادی و عبور از سد خونی-مغزی (BBB)
در محیط، سلولهای B به پلاسمابلاستهای ترشحکننده AQP4-IgG تمایز مییابند. IL-6 این تمایز را تسهیل کرده و نفوذپذیری BBB را افزایش میدهد. ناحیه postrema فاقد BBB است و میتواند مسیری برای ورود AQP4-IgG به CNS باشد.
زنجیره آسیب آستروسیتی
AQP4-IgG به کانالهای آبی AQP4 که با تراکم بالا در پایانههای آستروسیتی بیان میشوند متصل شده و از طریق مسیرهای زیر باعث آسیب آستروسیتی میشود.
فعال شدن مسیر کلاسیک کمپلمان: بخش Fc AQP4-IgG کمپلمان را فعال کرده و کمپلکس حمله غشایی (MAC) را تشکیل میدهد که مستقیماً به آستروسیتها آسیب میزند
ADCC (سمیت سلولی وابسته به آنتیبادی): سلولهای NK و نوتروفیلها از طریق گیرنده Fcγ به آستروسیتها آسیب میرسانند
رهاسازی آنافیلاتوکسین C5a: گرانولوسیتها (نوتروفیلها و ائوزینوفیلها) را جذب کرده و باعث آسیب ثانویه آکسون و دمیلیناسیون میشود1)
در MS، سلولهای T CD8+ نقش اصلی را ایفا میکنند و دمیلیناسیون عمدتاً در ماده سفید رخ میدهد، در حالی که در NMOSD، سلولهای T CD4+ نقش بیشتری دارند و ضایعات نکروتیک در هر دو ماده خاکستری و سفید ایجاد میشود.
دلیل توزیع
کانالهای AQP4 به وفور در عصب بینایی، ناحیه پوسترما و نخاع یافت میشوند و این نواحی به طور انتخابی هدف قرار میگیرند.
بیومارکرها
GFAP سرم: نشاندهنده آسیب آستروسیتها است و در هنگام حملات افزایش مییابد
زنجیره سبک نوروفیلامنت سرم (NfL): نشاندهنده آسیب آکسون است و با شدت حملات همبستگی دارد1)
سیتوکاینهای دخیل شامل IL-6، IL-10، IL-17a، G-CSF، TNF-α و BAFF/APRIL گزارش شدهاند.
7. آخرین تحقیقات و چشماندازهای آینده (گزارشهای مرحله تحقیقاتی)
تخمین زده میشود که ۳ تا ۵ درصد موارد NMOSD پارانئوپلاستیک باشند. موارد مرتبط با تراتوم تخمدان به طور ویژه به تفصیل مطالعه شدهاند.
Ikeguchi و همکاران (2021) مروری بر ۶ مورد NMOSD مرتبط با تراتوم تخمدان AQP4+ انجام دادند2). همه موارد زن، میانگین سن شروع ۳۲.۷ سال (۱۵-۵۰ سال). از ۶ مورد، ۸۳٪ (۵/۶ مورد) تهوع و استفراغ داشتند، ۸۳٪ باند الیگوکلونال CSF مثبت، ۸۳٪ ضایعات ساقه مغز پشتی داشتند. آنالیز پاتولوژی واکنش ایمنی AQP4 و نفوذ لنفوسیتی را در بافت عصبی GFAP مثبت داخل تومور تأیید کرد که نشاندهنده مکانیسم ارائه آنتیژن AQP4 در تومور و ایجاد پاسخ خودایمنی است. پس از برداشتن تومور، در ۶۰٪ (۳/۵ مورد) AQP4-IgG منفی شد.
Ding و همکاران (2021) مروری بر 43 مورد NMOSD پارانئوپلاستیک انجام دادند3). 88.4% از بیماران زن بودند و سرطان پستان و ریه شایعترین انواع تومور بودند. بهویژه در بیماران NMOSD بالای 50 سال، اهمیت غربالگری تومور تأکید شده است.
غربالگری تومورها از جمله تراتوم حتی در بیماران جوان توصیه میشود.
اثربخشی درمان با ایمونوجذب پروتئین A (IA) برای NMOSD مقاوم به درمان که به استروئیدها، پلاسمافرز و ریتوکسیماب پاسخ نمیدهد، گزارش شده است.
Fan و همکاران (2024) برای یک زن 35 ساله مبتلا به NMOSD مقاوم به درمان همراه با سندرم شوگرن که به پالس استروئید و IVIG پاسخ نداده بود، سه جلسه ایمونوجذب با پروتئین A انجام دادند 4). در عرض یک هفته، اختلال بینایی، پاراپلژی و اختلال حس عمقی به طور قابل توجهی بهبود یافت و کاهش سریع AQP4-IgG، IgA، IgG و IgM تأیید شد. در طول چهار سال پیگیری، عود یا پیشرفت بیماری مشاهده نشد.
واقعیت همراهی NMOSD با انواع بیماریهای خودایمنی در حال روشنتر شدن است.
Zhu و همکاران (2025) مورد یک دختر 14 ساله را گزارش کردند که در سن 11 سالگی به NMOSD مبتلا شده بود5). AQP4-IgG مثبت بود و در طول دوره، ابتلا به سندرم شوگرن اولیه تأیید شد. با تغییر درمان از متیلپردنیزولون، IVIG و مایکوفنولات موفتیل (MMF) به تاکرولیموس، بهبودی حفظ شده است. اگرچه در 20 تا 30 درصد موارد NMOSD بزرگسالان، همراهی با بیماریهای خودایمنی گزارش شده است، این مورد نشان میدهد که چنین همراهی در کودکان نیز وجود دارد5).
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, et al. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD: A case report and review. Brain Behav. 2021;11(9):e2282.
Fan W, Chen X, Xiao P, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD: a case report and literature review. Front Immunol. 2024;15:1429405.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.