رتینیت پیگمانتوزا (Retinitis Pigmentosa; RP) یک اصطلاح کلی برای گروهی از بیماریهای ارثی است که با دژنراسیون پیشرونده و گسترده سلولهای بینایی (میلهای و مخروطی) و اپیتلیوم رنگدانهدار شبکیه (RPE) مشخص میشود. دژنراسیون میلهای ابتدا رخ میدهد و سپس مخروطیها درگیر میشوند که به آن دیستروفی میلهای-مخروطی گفته میشود و RP مترادف با آن در نظر گرفته میشود. این یک بیماری واحد نیست بلکه گروهی از بیماریها با بیش از ۱۰۰ ژن درگیر است.
شیوع آن ۱ در ۴۰۰۰ تا ۸۰۰۰ نفر است و تعداد کل بیماران در ژاپن حداقل ۳۰٬۰۰۰ نفر تخمین زده میشود (۲۰٬۶۸۷ نفر دریافتکننده کمک هزینه بیماری نادر در سال ۲۰۲۳) 9). از نظر ارتباط با نابینایی، دومین علت نابینایی (۱۳.۰٪) در میان دریافتکنندگان جدید کارت معلولیت جسمی بالای ۱۸ سال (پس از آب سیاه با ۴۰.۷٪) و اولین علت نابینایی مادرزادی است 9). در ژاپن، این بیماری به عنوان بیماری نادر تعیینشده تحت قانون بیماریهای نادر (از ۱ ژانویه ۲۰۱۵) شناخته شده است 9) و مشمول کمک هزینه درمانی است.
همچنین، تمایز از سندرمهای متنوعی مانند PHARC (پلینوروپاتی، کمشنوایی، آتاکسی، RP، آب مروارید)، PCARP و سندرم الیور-مکفارلین نیز مهم است3).
Qآیا رتینیت پیگمانتوزا ارثی است؟
A
RP یک بیماری ارثی است، اما لزوماً به همه افراد منتقل نمیشود. خطر ارث بردن برای فرزندان بسته به الگوی وراثت متفاوت است. در نوع اتوزومال غالب، ۵۰٪ احتمال انتقال به فرزندان وجود دارد، اما در نوع اتوزومال مغلوب یا وابسته به X، خطر بسته به الگوی وراثت تغییر میکند. در موارد پراکنده (۴۸ تا ۶۳٪ از کل)، خطر ارث بردن برای نسل بعد معمولاً نسبتاً کم است9). استفاده از مشاوره ژنتیک توصیه میشود.
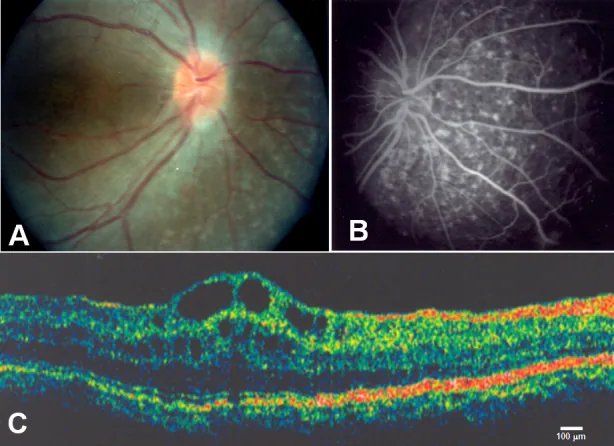
Zenteno JC, et al. Compound heterozygosity for a novel and a recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis. 2009. Figure 1. PMCID: PMC2742641. License: CC BY.
A: دروزنعصب بینایی و آتروفی گسترده اپیتلیوم رنگدانه شبکیه، B: فلورسانس عبوری مشیمیهای متناظر با آتروفی اپیتلیوم رنگدانه شبکیه، C: ادم ماکولای کیستیک و جداشدگی لایههای داخل شبکیه در ناحیه فووآ. مربوط به ادم ماکولای کیستیک است که در بخش «۲. علائم اصلی و یافتههای بالینی» بحث شده است.
علائم RP با توجه به مرحله پیشرفت تغییر میکند. از آنجایی که سلولهای استوانهای زودتر دچار انحطاط میشوند، شبکوری به عنوان اولین علامت اولیه ظاهر میشود.
شبکوری: کاهش دید یا دشواری در دیدن در مکانهای تاریک. به دلیل انحطاط اولیه سلولهای استوانهای از همان مراحل اولیه ظاهر میشود9)؛ معمولاً در دهه دوم تا سوم زندگی به صورت دشواری در دیدن در نور کم احساس میشود. در مراحل اولیه، عملکرد بینایی در روز اغلب طبیعی است.
تنگی میدان بینایی: میدان بینایی از محیط به سمت مرکز به تدریج باریک میشود. از اسکوتوم حلقوی به سمت تنگی متحدالمرکز (میدان بینایی تونلی) پیشرفت میکند9).
کاهش دید مرکزی: با پیشرفت انحطاط مخروطها به دنبال استوانهها، دید مرکزی نیز کاهش مییابد. در صورت وجود CME، ممکن است کاهش دید متوسط نسبتاً زودرس رخ دهد. در برخی موارد، دید مرکزی تا مراحل پایانی حفظ میشود.
فوتوفوبیا (کوری روزانه): حساسیت به نور. نشانه اختلال عملکرد مخروطها. با پیشرفت انحطاط مخروطها تشدید میشود. تمایز از پراکندگی نور ناشی از آب مروارید مهم است.
فوتوپسیا: ممکن است به دنبال انحطاط و از دست رفتن سلولهای گیرنده نور رخ دهد.
توهمات بینایی (سندرم شارل بونه): در بیمارانی که کاهش بینایی پیشرفته دارند، پدیده دیدن مناظر یا افرادی که واقعاً وجود ندارند. این یک تجربه پاتولوژیک نیست، بلکه پدیدهای ناشی از فعالیت بیش از حد قشر بینایی است9)
راهنمای پیشرفت علائم بر اساس مرحله بیماری در زیر آورده شده است.
آب مروارید زیر کپسول خلفی (PSC): در حدود 50% موارد رخ میدهد. کاهش دید در نور روشن مشخصه آن است. اگر عرض EZ (نوار بیضیشکل) 600 میکرومتر یا بیشتر باشد، دید خوب پس از جراحی آب مروارید قابل پیشبینی است (AUC 0.97)5)
ادم ماکولای کیستیک (CME): در 10 تا 50% موارد رخ میدهد و علت اصلی کاهش دید مرکزی است9)
گلوکوم زاویه بسته: در حدود 1% موارد حمله گزارش شده است؛ به دلیل ضعف زونولهای Zinn، سابلوکساسیون عدسی نیز ممکن است رخ دهد9)
غشای اپیرتینال (غشای پیشماکولار): در 15.6 تا 27.3% موارد رخ میدهد9)
سوراخ ماکولا و اسکیز حفرهای: نسبتاً نادر است، اما ممکن است نیاز به ویترکتومی داشته باشد9)
Qآیا پس از جراحی آب مروارید دید بهبود مییابد؟
A
در جراحی آب مروارید برای آب مروارید زیر کپسول خلفی همراه با RP، اگر عرض EZ در OCT قبل از عمل 600 میکرومتر یا بیشتر باشد، دید خوب پس از عمل قابل انتظار است (AUC 0.97)5). عرض EZ یک بیومارکر مفید برای پیشبینی عملکرد بینایی قبل از عمل است. با این حال، زونولهای Zinn اغلب ضعیف هستند و باید مراقب انقباض کپسول قدامی و دررفتگی IOL بود. برای پیشگیری از CME پس از عمل (10-14%)، استفاده طولانیتر از حد معمول از قطرههای استروئید و NSAID توصیه میشود9).
RP گروهی از بیماریها با ناهمگنی ژنتیکی بالا است که بیش از 100 ژن مختلف میتوانند عامل آن باشند9). ژنهای اصلی عامل در جمعیت ژاپنی بر اساس الگوی توارث در زیر آورده شده است.
مقایسه ژنهای اصلی عامل در زیر نشان داده شده است.
ژن
الگوی توارث
فراوانی/ویژگی در جمعیت ژاپنی
EYS
AR
30-50% موارد با ژن عامل شناساییشده (شایعترین در نوع AR)12)13)
USH2A
AR
دومین ژن شایع در نوع AR (4-9%)؛ ژن اصلی سندرم آشر12)
RHO
AD
شایعترین ژن در نوع AD 6)
RPGR
وابسته به X
حدود 70-75% در نوع XL 6)
REEP6
AR
یکی از ژنهای عامل نوع AR 4)
ویژگیهای هر ژن در زیر توضیح داده شده است.
EYS (Eyes Shut Homolog): شایعترین ژن عامل RP نوع AR در ژاپنیها (30-50% موارد شناسایی شده) 12)13). در اروپا و آمریکا به این فراوانی نیست و زمینه ژنتیکی خاص ژاپنیها را منعکس میکند.
USH2A: ژن اصلی سندرم آشر (RP + کمشنوایی) و دومین ژن شایع در RP نوع AR در ژاپنیها پس از EYS (4-9%) 12)
RHO (رودوپسین): شایعترین ژن عامل RP از نوع AD6). این ژن پروتئین گیرنده نور در سلولهای استوانهای شبکیه را کد میکند.
RPGR (تنظیمکننده GTPase رتینیت پیگمنتوزا): ژن اصلی عامل RP از نوع XL6). در بیماران مرد با جهش RPGR، مواردی از نارسایی مژک اولیه (PCD) گزارش شده است1).
REEP6 (پروتئین 6 افزایشدهنده بیان گیرنده): یکی از ژنهای عامل RP از نوع AR4).
نرخ تشخیص ژنهای عامل در آزمایش ژنتیک بسته به الگوی وراثت متفاوت است. برای نوع AD 35-60٪، برای نوع AR و موارد پراکنده 30-50٪، و برای نوع XL 16-36٪ گزارش شده است6).
همچنین، در RP سندرمیک شامل سندرم ژوبرت، سندرم باردت-بیدل و غیره، جهش در ژنهای مرتبط با مژک شایع است و ممکن است با عوارض سیستمیک (بیماری کلیوی، پلیداکتیلی، چاقی و غیره) همراه باشد2). افتراق از سندرمهای متنوعی مانند آتاکسی فردریش (PHARC)، PCARP، و سندرم الیور-مکفارلین نیز مهم است3).
Qآیا باید آزمایش ژنتیک انجام داد؟
A
تشخیص ژنتیکی در تشخیص قطعی، مشاوره ژنتیک و تعیین مناسب بودن درمان ژنی اهمیت دارد. سیستم پانل PrismGuide IRD (که به طور جامع توالی اگزونهای 82 ژن عامل IRD را تحلیل میکند) از سال 2023 تحت پوشش بیمه قرار گرفته است، اما تا ژوئن 2025 فقط برای افراد جوان با مشکوک به IRD مرتبط با RPE65 قابل استفاده است9). انجام آن همراه با مشاوره ژنتیک توصیه میشود. حتی بدون انجام تشخیص ژنتیک نیز میتوان مشاوره ژنتیک دریافت کرد.
حلقه فلورسانس غیرطبیعی (AF ring) به عنوان شاخص مرحله بیماری6)
تست میدان بینایی
ارزیابی پیشرفت
پریمتر Goldmann؛ برنامه HFA 10-2 نیز مفید است9)10)
تست دید رنگ
ارزیابی عملکرد مخروطها
ناهنجاری اکتسابی آبی-زرد شایع؛ تست پانل D-15 و 100 hue9)
تست تطابق با تاریکی
ارزیابی عملکرد استوانهها
نقطه عطف Kohlrausch تشخیص داده نمیشود10)
آزمایش ژنتیکی NGS
تشخیص ژنتیکی
پنل PrismGuide IRD (82 ژن) 9)
جزئیات هر آزمایش در زیر آورده شده است.
الکترورتینوگرافی (ERG): برای تشخیص قطعی ضروری است 6)9). از مراحل اولیه، پاسخ میلهای (ERG تیره) کاهش مییابد و با پیشرفت، پاسخ مخروطی (ERG روشن) نیز کاهش مییابد. الکترورتینوگرافی میدان کامل استاندارد است. اغلب در زمان مراجعه، غیرقابل ثبت است.
OCT (توموگرافی انسجام نوری): عرض و الگوی ناپدید شدن EZ (ناحیه بیضیشکل) ارزیابی میشود. عرض EZ به عنوان یک نشانگر زیستی کمی برای عملکرد بینایی و پیشآگهی مفید است و همچنین در تصمیمگیری برای جراحی آب مروارید استفاده میشود 5). از مراحل اولیه، نازک شدن لایه هستهای خارجی و ناپدید شدن EZ مشاهده میشود.
خودفلورسانس فوندوس (FAF): یک حلقه بیشفلورسانس غیرطبیعی (حلقه خودفلورسانس؛ AF ring) در اطراف ماکولا ظاهر میشود که نشاندهنده شبکیه با عملکرد باقیمانده و شاخص پیشرفت بیماری است 6).
تست میدان بینایی: تست میدان بینایی دینامیک با پریمتر Goldmann استاندارد است. با پیشرفت، اسکوتوم حلقوی به سمت تنگی میدان بینایی هممرکز پیش میرود 10). برنامه 10-2 Humphrey Field Analyzer (HFA) برای ارزیابی عملکرد مخروطی مرکزی باقیمانده مفید است 9).
تست دید رنگی: نقص آبی-زرد اکتسابی با فراوانی بالا مشاهده میشود. با تست Panel D-15 و 100 Hue ارزیابی میشود 9).
تست تطابق با تاریکی: نقطه عطف Kohlrausch (نقطه تغییر از میله به مخروط) تشخیص داده نمیشود 10).
توالییابی نسل بعدی (NGS): با سیستم پنل PrismGuide IRD، توالی اگزونهای 82 ژن بیماریزا به طور جامع تجزیه و تحلیل میشود 9). برای تعیین صلاحیت ژن درمانی نیز ضروری است.
در حال حاضر درمان قطعی برای RP وجود ندارد6)9). درمان بر حفظ عملکرد بینایی، مدیریت عوارض و حمایت از زندگی اجتماعی متمرکز است.
محافظت از سلولهای بینایی
ویتامین A (15000 IU/day): گزارش شده است که مصرف خوراکی آن پیشرفت ERG را چند درصد کند میکند14). هیچ تأثیری بر بهبود دید یا میدان بینایی ندارد. در مصرف طولانیمدت، نظارت بر عملکرد کبد ضروری است. در بارداری به دلیل خطر ناهنجاریهای جنینی منع مصرف دارد. در جهش ABCA4 ممکن است پیشرفت بیماری را تسریع کند14). توجه داشته باشید که ویتامین E ممکن است پیشرفت را تسریع کند و نیاز به احتیاط دارد14).
قطره چشمی اونوپروستون: تمایل به بهبود حساسیت وابسته به دوز مشاهده شد، اما در معیار اصلی فاز 2 (حساسیت شبکیه در 2 درجه مرکزی) تفاوت معنیدار نبود16).
نیلوا دیپین (مسدودکننده کانال کلسیم): یک گزارش طولانیمدت نشان داد که سرعت پیشرفت نقص میدان بینایی کاهش یافته است15). این گزارش از یک مرکز و با تعداد کمی از بیماران است و آزمایش چندمرکزی انجام نشده است.
N-استیل سیستئین (NAC): مهار استرس اکسیداتیو. در فاز I بهبود بینایی گزارش شده است17) و تا سال 2025 فاز III در حال انجام است.
DHA و لوتئین: از سلولهای بینایی ماکولا در برابر استرس اکسیداتیو محافظت میکنند. اثر اضافی DHA به ویتامین A تأیید نشده است.
هلنین (آداپتینول): برای بهبود موقت میدان بینایی و تطابق با تاریکی در RP تأیید شده است. ارزیابی اثربخشی در سطح پزشکی مدرن انجام نشده است.
عینک محافظ نور: استرس اکسیداتیو ناشی از اشعه UV و نور شدید را کاهش میدهد. استفاده روزانه توصیه میشود.
خط اول درمان، مهارکنندههای کربنیک آنهیدراز (CAI) است.
از قطره دورزولامید (تروسوپت) یا استازولامید خوراکی (دیاموکس) استفاده میشود. بهبود CMT در حدود 40% موارد حاصل میشود. حدود 30% عود دارند9).
داروهای ضد VEGF در CME ناشی از RP توصیه نمیشوند زیرا تولید VEGF کاهش یافته است9).
توجه داشته باشید که هیچکدام از این موارد برای RP-CME تأیید بیمه نشدهاند و استفاده خارج از برچسب محسوب میشوند.
جراحی آب مروارید: در موارد همراه با کاتاراکت زیرکپسولی خلفی انجام میشود. پهنای EZ ≥600 میکرومتر در OCT قبل از عمل، پیشبینیکننده دید خوب پس از جراحی است 5). در موارد ضعف زونولهای زین، استفاده از حلقه کشش کپسول عدسی را در نظر بگیرید. برای پیشگیری از CME پس از جراحی (10-14%)، از قطرههای استروئید و NSAIDs برای مدت طولانیتر از معمول استفاده کنید 9).
گلوکوم زاویه بسته: خطر ابتلا به گلوکوم زاویه بسته اولیه در بیماران RP بالاست. اتاق قدامی به تدریج کم عمق میشود؛ به صورت پیشگیرانه ایریدوتومی لیزری یا جراحی آب مروارید انجام دهید 9).
اپیرتینال ممبران (GL2026 CQ4): ویترکتومی. در مواردی که خط EZ پیوسته است، بهبود بینایی قابل انتظار است. در موارد ناپیوستگی خط EZ، بهبود محدود است. گزارشهایی از آتروفی شدید ماکولا در طولانی مدت پس از جراحی وجود دارد؛ بررسی در مراکز تخصصی توصیه میشود 9).
سوراخ ماکولا: ویترکتومی تنها درمان قطعی است. بررسی نتایج پس از جراحی محدود است 9).
حمایت و توانبخشی
مراقبت کمبینایی: دید کم ← ذرهبین، ذرهبین خوانا، تبلت؛ حساسیت به نور ← عینک تیره؛ تنگ شدن میدان بینایی ← عصای سفید؛ دید دور ← تکچشمی؛ عینک دید در شب. حمایت فردی متناسب با میدان بینایی و دید اهمیت دارد. استفاده از Smart Site (معرفی مراکز مشاوره کمبینایی در هر منطقه) توصیه میشود.
مشاوره ژنتیک: توسط متخصص ژنتیک بالینی و مشاور ژنتیک معتبر انجام میشود. تخمین خطر عود، مشاوره در مورد تحصیل، اشتغال، ازدواج و بارداری رایج است. حتی بدون انجام تشخیص ژنتیکی نیز میتوان مشاوره ژنتیک دریافت کرد.
سیستم بیماریهای نادر: به عنوان بیماری نادر معین، امکان استفاده از سیستم کمک هزینه درمانی وجود دارد 9). دریافت کارت معلولیت و حمایت پزشکی مستقل نیز در نظر گرفته شود.
وورتیژن نپارووک (LUXTURNA): داروی ژن درمانی قابل تجویز برای بیماران دارای واریانتهای بیماریزای دو آللی در ژن RPE65 و سلولهای شبکیه زنده کافی. در سال 2023 در ژاپن تأیید شد 9). در فاز III ایالات متحده (مطالعه 301)، 31 نفر ثبت نام کردند و در تحلیل mITT (20 نفر مداخله، 9 نفر کنترل)، MLMT و FST نور سفید به طور معنیداری در گروه مداخله بهبود یافت 18). در فاز III داخلی (مطالعه A11301) نیز در 4 بیمار ژاپنی افزایش حساسیت FST و گسترش میدان بینایی تأیید شد 19). اثر بهبود بینایی ضعیف است و آتروفی کوریورتینال به عنوان عارضه طولانی مدت در بیش از 20% گزارش شده است 9).
هر 6 ماه تا 1 سال موارد زیر انجام شود: دید، میکروسکوپ اسلیت لمپ، فوندوس، Humphrey میدان بینایی (HFA 10-2)، OCT9).
Qبرای درمان ادم ماکولا از چه داروهایی استفاده میشود؟
A
خط اول درمان RP-CME مهارکنندههای کربنیک آنهیدراز (CAI) است و از قطره دورزولامید یا استازولامید خوراکی استفاده میشود 9). بهبود CMT در حدود 40% موارد حاصل میشود، اما حدود 30% عود دارند. در صورت عدم پاسخ به CAI، تزریق داخل زجاجیهای تریامسینولون استونید یا ایمپلنت داخل زجاجیهای دگزامتازون (Ozurdex) گزینههای بعدی هستند. داروهای ضد VEGF در RP-CME توصیه نمیشوند. توجه به این نکته ضروری است که همه این موارد استفاده خارج از برچسب هستند.
در RP ابتدا سلولهای بینایی میلهای تخریب و ناپدید میشوند و سپس سلولهای مخروطی به صورت ثانویه تخریب میشوند 7). مخروطها برای بقا به فاکتورهای تغذیهای تولید شده توسط میلهها (RdCVF) وابسته هستند، بنابراین پس از ناپدید شدن میلهها، مخروطها نیز عملکرد خود را از دست میدهند 7)11).
شبکیه یکی از بافتهای با بیشترین فعالیت متابولیک است و 80 تا 90% گلوکز را از طریق گلیکولیز هوازی (اثر Warburg) به لاکتات تبدیل میکند. مخروطها نسبت به میلهها در برابر استرس متابولیک آسیبپذیرتر هستند و این آسیبپذیری متابولیک نیز به تخریب ثانویه مخروطها کمک میکند 11).
التهاب نیز به عنوان یک عامل اصلی در پیشرفت RP شناخته میشود و فعال شدن میکروگلیا و نفوذ ماکروفاژها آسیب شبکیه را تشدید میکند 11). استرس اکسیداتیو نیز به عنوان یک محرک بیولوژیک برای تخریب ثانویه مخروطها عمل میکند.
جهش RHO: رودوپسین با تا شدن نادرست باعث استرس شبکه آندوپلاسمی → پاسخ پروتئین تا نشده (UPR) → آپوپتوز میشود 11)
جهش REEP6: REEP6 پروتئینی را کد میکند که در حفظ مورفولوژی شبکه آندوپلاسمی (ER) نقش دارد. جهشهای بیماریزا منجر به تشکیل اجسام انکلوژن در ER در بخش خارجی میلهها شده و به دژنراسیون سلولهای بینایی میانجامد4)
جهش RPGR: RPGR در ساختار اکسونمال مژک اولیه نقش دارد و جهش آن انتقال مواد به بخش خارجی گیرنده نوری را مختل میکند1)
7. تحقیقات جدید و چشماندازهای آینده (گزارشهای در مرحله تحقیق)
ژن درمانی امیدوارکنندهترین رویکرد برای درمان بیماریهای ارثی شبکیه است8).
Luxturna (voretigene neparvovec): یک داروی ژن درمانی قابل تجویز برای بیماران دارای واریانتهای بیماریزا در هر دو آلل ژن RPE65. در فاز III ایالات متحده (مطالعه 301)، 31 نفر ثبتنام کردند و در آنالیز mITT (20 نفر مداخله، 9 نفر کنترل)، MLMT و FST نور سفید به طور معنیداری در گروه مداخله بهبود یافت18). در فاز III داخلی (مطالعه A11301) نیز در 4 بیمار ژاپنی افزایش حساسیت FST و گسترش میدان بینایی تأیید شده است19). این دارو در سال 2023 در ژاپن تأیید شده و به عنوان پل ارتباطی بین درمان استاندارد و تحقیقاتی محسوب میشود.
ژن درمانی RPGR: ژن درمانی با واسطه AAV برای RP نوع وابسته به X ناشی از جهش RPGR تا کارآزماییهای بالینی فاز I/II/III پیش رفته است8).
CRISPR/Cas9: تحقیقات برای اصلاح مستقیم جهشهای بیماریزا یا غیرفعالسازی جهشهای غالب منفی در حال انجام است8).
RdCVF (عامل بقای مخروطی مشتق از میله) و درمان محافظت از مخروطها
RdCVF پروتئینی است که از میلهها ترشح شده و بقای مخروطها را حفظ میکند7)11). کارآزماییهای بالینی درمان محافظت از مخروطها با استفاده از RdCVF در حال انجام است و به عنوان یک استراتژی درمانی مستقل برای حفظ عملکرد مخروطها پس از دژنراسیون میلهها مورد توجه قرار گرفته است.
مطالعات in vivo اخیر (مدل موش rd10) نشان داده است که تزریق داخل زجاجیهای دگزامتازون از سلولهای مخروطی و اپیتلیوم رنگدانهای شبکیه محافظت میکند 11). گلوکوکورتیکوئیدها پتانسیل بالایی برای استفاده مجدد به عنوان درمانهای مستقل از جهش دارند. با این حال، شواهد فعلی محدود به مدلهای حیوانی است و برای کاربرد بالینی در انسان به تأیید بیشتری نیاز است.
پیوند شبکیه مشتق از سلولهای iPS: تحقیقات در مورد پیوند لایههای سلولهای بینایی ساخته شده از سلولهای iPS خود بیمار در حال انجام است.
شبکیه مصنوعی (پروتز شبکیه): دستگاههای تحریک الکتریکی برای RP مرحله نهایی. Argus II و دیگران در خارج از کشور عملیاتی شدهاند و در ژاپن، کارآزمایی بالینی روش تحریک از طریق اپیکوروئید در حال انجام است.
Qآیا ژن درمانی در ژاپن نیز در دسترس است؟
A
وورتیژن نپارووک (لوکستورنا) در سال 2023 در ژاپن تأیید شد، اما فقط برای دیستروفی شبکیه با واریانتهای بیماریزای دو آللی در ژن RPE65 محدود است 9)18)19). ژن درمانی برای RP با سایر جهشهای ژنی از جمله جهش RPGR در مرحله کارآزمایی بالینی است 7) و به عنوان یک درمان عمومی در ژاپن تأیید نشده است.
Qبرای دریافت درمانهای تحقیقاتی چه باید کرد؟
A
شرکت در کارآزماییهای بالینی فقط به آزمایشهای رسمی که توسط کمیته اخلاق مؤسسه پزشکی تأیید شده است محدود میشود. علاوه بر مشورت با پزشک معالج، میتوانید اطلاعات کارآزمایی را در پایگاه اطلاعات کارآزمایی بالینی (jRCT) که توسط مرکز ملی سرطان اداره میشود و clinicaltrials.gov ایالات متحده جستجو کنید.
Baz-Redón N, Sánchez-Bellver L, Fernández-Cancio M, Rovira-Amigo S, Burgoyne T, Ranjit R, Aquino V, Toro-Barrios N, et al. Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene. Cells. 2024;13(6). doi:10.3390/cells13060524. PMID:38534367; PMCID:PMC10968961.
Holanda IP, et al. Syndromic Retinitis Pigmentosa: A 15-Patient Study Exploring Clinical and Genetic Features. Genes. 2024;15(4):516.
Wawrocka A, Walczak-Sztulpa J, Kuszel L, Niedziela-Schwartz Z, Skorczyk-Werner A, Bernardczyk-Meller J, Krawczynski MR. Coexistence of Retinitis Pigmentosa and Ataxia in Patients with PHARC, PCARP, and Oliver-McFarlane Syndromes. International journal of molecular sciences. 2024;25(11). doi:10.3390/ijms25115759. PMID:38891946; PMCID:PMC11172263.
Lujia Zhang, Ya Li, Litao Qin, Yu Wu, Bo Lei. Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population. Genes. 2021;12(4):537. doi:10.3390/genes12040537.
Daiki Sakai, Seiji Takagi, Yasuhiko Hirami, Makoto Nakamura, Yasuo Kurimoto. Use of ellipsoid zone width for predicting visual prognosis after cataract surgery in patients with retinitis pigmentosa. Eye. 2022;37(1):42-47. doi:10.1038/s41433-021-01878-3.
日本眼科学会. 網膜色素変性診療ガイドライン. 日眼会誌. 2016;120:846-861.
Campochiaro PA, Mir TA. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res. 2018;62:24-37. doi:10.1016/j.preteyeres.2017.08.004.
Liu Y, Zong X, Cao W, Zhang W, Zhang N, Yang N. Gene therapy for retinitis pigmentosa: current challenges and new progress. Biomolecules. 2024;14(8):903. PMID: 39199291. PMCID: PMC11352491. doi:10.3390/biom14080903.
Napoli D, Di Marco B, Salamone G, Orsini N, Mazziotti R, Strettoi E. Keeping the lights on: a new role for an old drug to support cone survival in Retinitis Pigmentosa. Progress in retinal and eye research. 2025;109:101403. doi:10.1016/j.preteyeres.2025.101403. PMID:40998215.
Koyanagi Y, Akiyama M, Nishiguchi KM, Momozawa Y, Kamatani Y, Takata S, et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. Journal of medical genetics. 2019;56(10):662-670. doi:10.1136/jmedgenet-2018-105691. PMID:31213501.
Numa S, Oishi A, Higasa K, Oishi M, Miyata M, Hasegawa T, Ikeda HO, Otsuka Y, Matsuda F, Tsujikawa A.. EYS is a major gene involved in retinitis pigmentosa in Japan: genetic landscapes revealed by stepwise genetic screening. Sci Rep. 2020;10(1):20770. doi:10.1038/s41598-020-77558-1. PMID:33247286; PMCID:PMC7695703.
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, Willett W.. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. doi:10.1001/archopht.1993.01090060049022. PMID:8512476.
Nakazawa M, Suzuki Y, Ito T, Metoki T, Kudo T, Ohguro H. Long-term effects of nilvadipine against progression of the central visual field defect in retinitis pigmentosa: an extended study. BioMed research international. 2013;2013:585729. doi:10.1155/2013/585729. PMID:24319686; PMCID:PMC3844269.
Yamamoto S, Sugawara T, Murakami A, Nakazawa M, Nao-I N, Machida S, et al. Topical isopropyl unoprostone for retinitis pigmentosa: microperimetric results of the phase 2 clinical study. Ophthalmology and therapy. 2012;1(1):5. doi:10.1007/s40123-012-0005-9. PMID:25135585; PMCID:PMC4108136.
Campochiaro PA, Iftikhar M, Hafiz G, Akhlaq A, Tsai G, Wehling D, et al. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. The Journal of clinical investigation. 2020;130(3):1527-1541. doi:10.1172/JCI132990. PMID:31805012; PMCID:PMC7269599.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.