Retinitis Pigmentosa (RP), fotoreseptörler (çubuk ve koni hücreleri) ve retina pigment epitelinde (RPE) ilerleyici yaygın dejenerasyon ile karakterize kalıtsal hastalık grubunun genel adıdır. Çubuk dejenerasyonu önce gelir ve koni dejenerasyonu daha sonra ortaya çıkar; buna çubuk-koni distrofisi denir ve RP ile eş anlamlı kabul edilir. Tek bir hastalık değil, 100’den fazla genin rol oynadığı bir hastalık grubudur.
Prevalansı 4.000-8.000’de 1’dir ve Japonya’da toplam hasta sayısı en az 30.000’i aşmaktadır (2023 yılı belirlenmiş nadir hastalık yardımı alan 20.687 kişi) 9). Görme engeli ile ilişkili olarak, 18 yaş üstü yeni fiziksel engelli raporu alanlarda görme engelinin ikinci en sık nedeni (%13,0; 2019 yılı, glokom %40,7’den sonra) ve konjenital körlüğün birinci nedenidir 9). Japonya’da Nadir Hastalıklar Yasası kapsamında belirlenmiş nadir hastalık (1 Ocak 2015’ten itibaren) olarak tanınmıştır 9) ve sağlık harcaması desteği kapsamındadır.
RP ayrıca diğer sistemik hastalıklarla birlikte görülen sendromik RP şeklinde de bulunur ve üst kavram olarak siliyopati altında aşağıdaki gibi sınıflandırılır9)2).
Siliyopati:
Usher sendromu (tip 1/2/3): RP + işitme kaybı; belirlenmiş nadir hastalık (AR). Tip 1’de erken çocukluktan itibaren şiddetli işitme kaybı ve vestibüler disfonksiyon eşlik eder.
Ayrıca, PHARC (polinöropati, işitme kaybı, ataksi, RP, katarakt), PCARP ve Oliver-McFarlane sendromu gibi çeşitli sendromlardan ayırt edilmesi de önemlidir3).
QRetinitis pigmentosa kalıtsal mıdır?
A
RP kalıtsal bir hastalıktır, ancak herkese mutlaka geçmez. Kalıtım şekline bağlı olarak çocuğa geçme riski değişir. Otozomal dominant tipte çocuğa geçme olasılığı %50 iken, otozomal resesif veya X’e bağlı tipte risk kalıtım şekline göre değişir. Sporadik vakalarda (toplamın %48-63’ü) sonraki nesle geçme riski genellikle daha düşüktür9). Genetik danışmanlık önerilir.
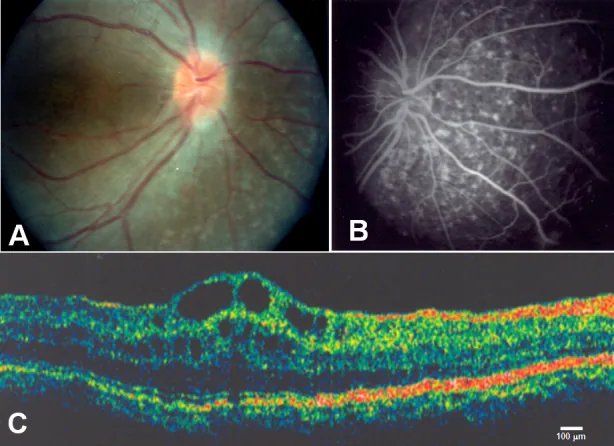
Zenteno JC, et al. Compound heterozygosity for a novel and a recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis. 2009. Figure 1. PMCID: PMC2742641. License: CC BY.
A: Optik sinir başı druseni ve yaygın retina pigment epiteli atrofisi, B: Retina pigment epiteli atrofisine karşılık gelen koroidal geçiş floresansı, C: Kistoid makula ödemi ve foveada retina iç tabakalarının ayrışması. Metnin “2. Başlıca belirtiler ve klinik bulgular” bölümünde ele alınan kistoid makula ödemine karşılık gelir.
RP’nin belirtileri ilerleme evresine göre değişir. Çubuk fotoreseptörler önce dejenerasyona uğradığından, en erken belirti olarak gece körlüğü ortaya çıkar.
Gece körlüğü: Karanlıkta görme azalması veya zorluğu. Çubuk fotoreseptörlerin öncelikli dejenerasyonu nedeniyle en erken evreden itibaren ortaya çıkar9); 10-20’li yaşlarda loş ortamlarda görme zorluğu olarak fark edilir. Erken dönemde gündüz görme işlevi genellikle normaldir.
Görme alanı daralması: Çevresel görme alanı içe doğru giderek daralır. Halka şeklindeki skotomdan santral daralmaya (tünel görüşü) ilerler9).
Görme keskinliğinde azalma: Çubukları takiben koni dejenerasyonu ilerledikçe merkezi görme de azalır. CME varlığında nispeten erken dönemde orta derecede görme azalması görülebilir. Bazı vakalarda merkezi görme son evreye kadar korunabilir.
Fotofobi (gündüz körlüğü): Işığa karşı hassasiyet. Koni işlev bozukluğunun bir belirtisidir. Koni dejenerasyonu ilerledikçe artar. Katarakta bağlı ışık saçılımından ayırt edilmesi önemlidir.
Fotopsi: Fotoreseptörlerin dejenerasyonu ve kaybına bağlı olarak ortaya çıkabilir.
Görsel halüsinasyonlar (Charles Bonnet sendromu): Görme keskinliği ileri derecede azalmış hastalarda, gerçekte var olmayan manzaralar veya kişiler görme olgusu. Patolojik bir deneyim değil, görsel korteksin aşırı aktivitesine bağlı bir fenomendir9)
Hastalık evrelerine göre semptom ilerlemesinin kılavuzu aşağıda verilmiştir.
Evre
Ana belirtiler
Yaklaşık zaman
Erken
Gece körlüğü
10-20’li yaşlar
Orta
Görme alanı daralması (halka şeklinde skotom → santral)
Arka subkapsüler katarakt (PSC): Yaklaşık %50’sinde görülür. Aydınlık ortamda görme azalması karakteristiktir. EZ (eliptik bölge) genişliği 600 μm veya üzerinde ise katarakt cerrahisi sonrası iyi görme öngörülebilir (AUC 0.97)5)
Kistoid makula ödemi (CME): %10-50’sinde görülür ve merkezi görme azalmasının ana nedenidir9)
Açı kapanması glokomu: Yaklaşık %1’inde atak bildirilmiştir; Zinn zonüllerinin zayıflığına bağlı lens subluksasyonu da oluşabilir9)
Makula deliği ve foveal şizis: Nispeten nadirdir ancak vitrektomi gerektirebilir9)
QKatarakt cerrahisi sonrası görme düzelir mi?
A
RP’ye eşlik eden arka subkapsüler katarakt için yapılan katarakt cerrahisinde, ameliyat öncesi OCT’de EZ genişliği 600 μm veya üzerinde ise iyi bir postoperatif görme beklenebilir (AUC 0.97)5). EZ genişliği, ameliyat öncesi görsel işlevi öngörmede yararlı bir biyobelirteçtir. Ancak Zinn zonülleri sıklıkla zayıftır ve ön kapsül kontraksiyonu ile IOL dislokasyonuna dikkat edilmelidir. Postoperatif CME’ye (%10-14) karşı steroid ve NSAID damlalarının normalden daha uzun süre kullanılması önerilir9).
RP, 100’den fazla gen mutasyonunun neden olduğu genetik heterojenitesi yüksek bir hastalık grubudur9). Japon popülasyonunda başlıca nedensel genler kalıtım şekline göre aşağıda gösterilmiştir.
Başlıca nedensel genlerin karşılaştırması aşağıda gösterilmiştir.
Gen
Kalıtım şekli
Japon popülasyonunda sıklık/özellik
EYS
AR
Nedensel geni tanımlanan olguların %30-50’si (AR tipte en sık)12)13)
USH2A
AR
AR tipinde ikinci en sık (%4-9); Usher sendromunun ana geni 12)
RHO
AD
AD tipinde en sık 6)
RPGR
X’e bağlı
XL tipinde yaklaşık %70-75 6)
REEP6
AR
AR tipinin neden olan genlerinden biri 4)
Her genin özellikleri aşağıda açıklanmıştır.
EYS (Eyes Shut Homolog): Japonlarda AR tip RP’nin en sık neden olan geni (tanımlanan vakaların %30-50’si) 12)13). Avrupa ve Amerika’da bu kadar sık değildir ve Japonlara özgü genetik arka planı yansıtır.
USH2A: Usher sendromunun (RP + işitme kaybı) ana geni ve Japonlarda AR tip RP’de EYS’den sonra ikinci en sık (%4-9) 12)
RHO (Rodopsin): AD tipi RP’nin en sık neden olan geni6). Çubuk fotoreseptör hücrelerindeki ışık alıcı proteini kodlar.
RPGR (Retinitis Pigmentosa GTPaz Düzenleyici): XL tipi RP’nin ana neden olan geni6). RPGR mutasyonu olan erkek hastalarda primer siliyer disfonksiyon (PCD) eşlik eden vakalar bildirilmiştir1).
REEP6 (Reseptör Ekspresyonunu Artıran Protein 6): AR tipi RP’nin neden olan genlerinden biridir4).
Genetik testte neden olan genlerin saptanma oranı kalıtım şekline göre değişir. AD tipinde %35-60, AR tipi ve sporadik vakalarda %30-50, XL tipinde %16-36 olduğu bildirilmiştir6).
Ayrıca, Joubert sendromu, Bardet-Biedl sendromu gibi sendromik RP’de silya ile ilişkili gen mutasyonları sıktır ve sistemik komplikasyonlar (böbrek hastalığı, polidaktili, obezite vb.) eşlik edebilir2). Friedreich ataksisi (PHARC), PCARP, Oliver-McFarlane sendromu gibi çeşitli sendromlardan ayırıcı tanı da önemlidir3).
QGenetik test yaptırmalı mıyım?
A
Genetik tanı, kesin tanı, genetik danışmanlık ve gen tedavisi uygunluğunun belirlenmesinde önemlidir. PrismGuide IRD Panel Sistemi (82 IRD nedensel geninin ekzon dizilerini kapsamlı olarak analiz eder) 2023 yılında sigorta kapsamına alınmıştır, ancak Haziran 2025 itibarıyla yalnızca RPE65 ile ilişkili IRD şüphesi olan genç başlangıçlı hastalar için geçerlidir9). Genetik danışmanlık ile birlikte yapılması önerilir. Genetik tanı yaptırmadan da genetik danışmanlık alınabilir.
Gece körlüğü, görme alanı daralması, görme azalması veya fotofobiden en az biri
B. Test bulguları (aşağıdakilerden en az ikisi):
(1) Fundus bulguları: Retina damarlarında incelme, pürüzlü retina rengi, kemik korpüskülü benzeri pigment birikimleri, çok sayıda beyaz nokta, optik atrofi veya makula dejenerasyonu
Edinsel mavi-sarı anomali sık; Panel D-15 ve 100 hue testi9)
Karanlığa uyum testi
Çubuk işlevi değerlendirmesi
Kohlrausch bükülme noktası saptanmaz10)
NGS gen testi
Genetik tanı
PrismGuide IRD paneli (82 gen) 9)
Her testin detayları aşağıda verilmiştir.
Elektroretinografi (ERG): Kesin tanı için gereklidir 6)9). Erken dönemde çubuk yanıtı (karanlık adaptasyon ERG) azalır, ilerledikçe koni yanıtı (aydınlık adaptasyon ERG) da azalır. Tam alan elektroretinografi standarttır. Başvuru sırasında genellikle kaydedilemez durumdadır.
OCT (Optik Koherens Tomografi): EZ (eliptik bölge) genişliği ve kaybolma paterni değerlendirilir. EZ genişliği, görsel fonksiyon ve prognoz için kantitatif bir biyobelirteç olarak faydalıdır ve katarakt cerrahisi kararında da kullanılır 5). Erken dönemden itibaren dış nükleer tabakada incelme ve EZ kaybı görülür.
Fundus otofloresansı (FAF): Makula çevresinde anormal hiperfloresan halka (otofloresan halka; AF halkası) ortaya çıkar ve kalan fonksiyonel retinayı gösteren hastalık ilerlemesinin bir göstergesidir 6).
Görme alanı testi: Goldmann perimetri ile dinamik görme alanı testi standarttır. İlerlemeyle birlikte halka şeklinde skotomdan santripetal görme alanı daralmasına doğru gider 10). Humphrey Görme Alanı (HFA) 10-2 programı, kalan santral koni fonksiyonunun değerlendirilmesinde faydalıdır 9).
Renk görme testi: Edinsel mavi-sarı renk körlüğü sık görülür. Panel D-15 testi ve 100 hue testi ile değerlendirilir 9).
Karanlık adaptasyon testi: Kohlrausch bükülme noktası (çubuk ve koni geçiş noktası) tespit edilemez 10).
Yeni nesil dizileme (NGS): PrismGuide IRD panel sistemi ile 82 hastalık geninin ekzon dizileri kapsamlı olarak analiz edilebilir 9). Gen tedavisi uygunluğunun belirlenmesinde de gereklidir.
Şu anda RP için kesin bir tedavi yoktur6)9). Tedavi, görme fonksiyonunun korunması, komplikasyonların yönetimi ve sosyal yaşam desteğine odaklanır.
Fotoreseptör Koruması
A Vitamini (15.000 IU/gün): Oral alımın ERG kötüleşmesini yüzde birkaç oranında geciktirdiği bildirilmiştir14). Görme keskinliği veya görme alanında iyileşme sağlamaz. Uzun süreli kullanımda karaciğer fonksiyon takibi gereklidir. Gebelikte teratojenik risk nedeniyle kontrendikedir. ABCA4 mutasyonlarında hastalığın ilerlemesini hızlandırabilir14). E vitamininin ilerlemeyi hızlandırabileceği için dikkatli olunmalıdır14).
Unoproston göz damlası: Doza bağlı hassasiyet iyileşme eğilimi gözlenmiş ancak Faz 2 çalışmasının birincil sonlanım noktasında (merkezi 2 derece retina hassasiyeti) anlamlı fark bulunmamıştır16).
Nilvadipin (kalsiyum kanal blokeri): Uzun dönemli bir raporda görme alanı kaybının ilerleme hızının yavaşladığı bildirilmiştir15). Bu rapor tek merkezli ve az sayıda hastayı içermektedir; çok merkezli doğrulama çalışması yapılmamıştır.
N-Asetil Sistein (NAC): Oksidatif stresi baskılar. Faz I çalışmasında görme keskinliğinde iyileşme bildirilmiş17) ve 2025 itibarıyla Faz III devam etmektedir.
DHA ve Lutein: Makula fotoreseptörlerini oksidatif strese karşı korur. DHA’nın A vitaminine ek etkisi doğrulanmamıştır.
Helenien (Adaptinol): RP’de geçici görme alanı ve karanlığa uyum iyileşmesi için onaylanmıştır. Modern tıp standartlarında etkinlik değerlendirmesi yapılmamıştır.
Işık koruyucu gözlük: UV ve parlak ışığa bağlı oksidatif stresi azaltır. Günlük kullanım önerilir.
Birinci basamak tedavi karbonik anhidraz inhibitörleridir (KAİ).
Dorzolamid (Trusopt) göz damlası veya asetazolamid (Diamox) oral kullanılır. CMT iyileşmesi yaklaşık %40 oranında sağlanır. Yaklaşık %30’unda nüks görülür9).
KAİ’ye dirençli durumlarda steroidler düşünülür.
Triamsinolon asetonid (Macuoid) intravitreal enjeksiyonu veya deksametazon intravitreal implantı (Ozurdex) kullanılır.
Anti-VEGF ilaçlar RP-KMÖ’de önerilmez çünkü VEGF üretimi azalmıştır9).
Hiçbirinin RP-CME için sigorta onayı olmadığına ve endikasyon dışı kullanım olduğuna dikkat edin.
Katarakt cerrahisi: Arka subkapsüler kataraktı olan vakalarda uygulanır. Ameliyat öncesi OCT’de EZ genişliği ≥600 μm, iyi postoperatif görme keskinliğinin öngördürücüsüdür 5). Zinn zonülleri zayıf olan vakalarda lens kapsül germe halkası kullanmayı düşünün. Postoperatif CME’yi (%10-14) önlemek için steroid ve NSAID damlaları normalden daha uzun süre kullanın 9).
Epiretinal membran (GL2026 CQ4): Vitrektomi. EZ çizgisi sürekli olan vakalarda görme iyileşmesi beklenebilir. EZ çizgisi süreksiz olan vakalarda iyileşme sınırlıdır. Uzun dönemde ciddi makula atrofisi bildirilmiştir; uzman merkezlerde değerlendirme önerilir 9).
Az görme bakımı: Düşük görme → büyüteç, büyüteçli okuma cihazı, tablet; fotofobi → güneş gözlüğü; görme alanı daralması → beyaz baston; uzak görme → monoküler; gece görüş destek gözlüğü. Görme alanı ve keskinliğine göre bireysel destek önemlidir. Smart Site (her bölgedeki az görme danışma merkezlerinin tanıtımı) kullanımı önerilir.
Genetik danışmanlık: Klinik genetik uzmanı ve sertifikalı genetik danışman tarafından yürütülür. Tekrarlama riski tahmini, eğitim, iş, evlilik ve doğumla ilgili danışmanlık sıktır. Genetik tanı alınmasa bile genetik danışma alınabilir.
Nadir hastalık sistemi: Belirlenmiş nadir hastalık olarak tıbbi masraf destek sisteminden yararlanılabilir 9). Engelli kimlik kartı ve bağımsız tıbbi destek alınması da değerlendirilir.
Voretigen neparvovek (LUXTURNA): RPE65 geninde biallelik patojenik varyantları olan ve yeterli canlı retina hücresine sahip hastalara uygulanabilen gen tedavisi ilacı. 2023’te Japonya’da onaylanmıştır 9). ABD Faz III (301 çalışması) 31 kişi kaydetmiş, mITT analizinde (20 müdahale, 9 kontrol) MLMT ve beyaz ışık FST, kontrol grubuna göre anlamlı iyileşme göstermiştir 18). Yerel Faz III (A11301 çalışması) da 4 Japon hastada FST duyarlılığında artış ve görme alanı genişlemesi doğrulanmıştır 19). Görme keskinliği iyileştirme etkisi zayıftır ve uzun dönem komplikasyon olarak %20’den fazla koroidoretinal atrofi bildirilmiştir 9).
6 ayda bir ila yılda bir şunlar yapılır: görme keskinliği, yarık lamba biyomikroskopisi, fundus, Humphrey görme alanı (HFA 10-2), OCT9).
QMakula ödeminin tedavisinde hangi ilaçlar kullanılır?
A
RP-CME’de birinci basamak tedavi karbonik anhidraz inhibitörleridir (CAI) ve dorzolamid damla veya asetazolamid oral olarak kullanılır 9). CMT’de iyileşme yaklaşık %40 oranında sağlanır, ancak yaklaşık %30’unda nüks görülür. CAI ile düzelme olmazsa, triamsinolon asetonid intravitreal enjeksiyonu veya deksametazon intravitreal implant (Ozurdex) seçeneklerdir. Anti-VEGF ilaçları RP-CME’de önerilmez. Tümünün sigorta kapsamı dışı kullanım olduğuna dikkat edilmelidir.
RP’de tipik olarak önce çubuk fotoreseptörler dejenere olur ve kaybolur, ardından koni fotoreseptörler sekonder olarak dejenere olur 7). Koniler, çubuklar tarafından üretilen besin faktörlerine (RdCVF) bağımlı olduğu için, çubuklar kaybolduktan sonra koniler de işlevlerini kaybeder 7)11).
Retina, metabolik olarak en aktif dokulardan biridir ve glikozun %80-90’ını aerobik glikoliz (Warburg etkisi) yoluyla laktata dönüştürür. Koniler, çubuklara göre metabolik strese karşı daha hassastır ve bu metabolik kırılganlık da sekonder koni dejenerasyonuna katkıda bulunur 11).
İnflamasyon da RP ilerlemesinde önemli bir faktör olarak kabul edilmektedir; mikroglia aktivasyonu ve makrofaj infiltrasyonu retina hasarını kötüleştirir 11). Oksidatif stres de sekonder koni dejenerasyonunun biyolojik bir itici gücü olarak işlev görür.
Gene bağlı olarak dejenerasyon mekanizması farklılık gösterir.
RHO mutasyonu: Yanlış katlanmış rodopsin, endoplazmik retikulum stresi → UPR (katlanmamış protein yanıtı) → apoptozu indükler 11)
REEP6 mutasyonu: REEP6, ER morfolojisinin korunmasında rol oynayan bir proteini kodlar. Patojenik mutasyonlar, çubuk dış segmentinde ER içi inklüzyon cisimciklerinin oluşumuna yol açar ve fotoreseptör dejenerasyonuna neden olur4)
RPGR mutasyonu: RPGR, birincil siliumun aksonem yapısında rol oynar ve mutasyon, fotoreseptör dış segmentine madde taşınmasını bozar1)
7. Güncel Araştırmalar ve Gelecek Perspektifleri (Araştırma Aşamasındaki Raporlar)
Gen tedavisi, kalıtsal retina hastalıklarının tedavisi için en umut verici yaklaşımdır8).
Luxturna (voretigene neparvovec): RPE65 geninin her iki allelinde patojenik varyantları olan hastalara uygulanabilen bir gen tedavisi ilacı. ABD Faz III (301 çalışması) 31 kişi kaydetti ve mITT analizinde (20 müdahale, 9 kontrol) MLMT ve beyaz ışık FST, kontrol grubuna göre anlamlı şekilde iyileşti18). Yerel Faz III (A11301 çalışması) ayrıca 4 Japon hastada FST duyarlılığında artış ve görme alanında genişleme olduğunu doğruladı19). 2023 yılında Japonya’da onaylanmış olup, standart tedavi ile araştırma tedavisi arasında bir köprü görevi görmektedir.
RPGR gen tedavisi: RPGR mutasyonuna bağlı XL tipi RP için AAV aracılı gen tedavisi, Faz I/II/III klinik çalışmalarına kadar ilerlemiştir8).
CRISPR/Cas9: Patojenik mutasyonların doğrudan düzeltilmesi veya dominant negatif mutasyonların inaktivasyonu için araştırmalar devam etmektedir8).
RdCVF (Çubuk Kaynaklı Konik Sağkalım Faktörü) ve Konik Koruma Tedavisi
RdCVF, çubuklardan salgılanan ve koniklerin sağkalımını sürdüren bir proteindir7)11). RdCVF kullanılarak yapılan konik koruma tedavisinin klinik çalışmaları devam etmektedir ve çubuk dejenerasyonu sonrası konik fonksiyonunu koruyan bağımsız bir tedavi stratejisi olarak dikkat çekmektedir.
NAC, oksidatif stresi baskılayan bir ilaçtır ve Faz I çalışmasında görme keskinliğinde iyileşme bildirilmiştir17). 2025 itibarıyla Faz III çalışması devam etmektedir.
Glukokortikoid (Deksametazon) Yeniden Kullanım Potansiyeli
Son in vivo çalışmalar (rd10 fare modeli), intravitreal deksametazonun koni fotoreseptörlerini ve retina pigment epitelini koruduğunu göstermiştir 11). Glukokortikoidler, mutasyondan bağımsız tedaviler olarak güçlü bir yeniden kullanım potansiyeline sahiptir. Ancak mevcut kanıtlar yalnızca hayvan modelleriyle sınırlıdır ve insanlarda klinik uygulama için daha fazla doğrulama gereklidir.
iPS Hücre Kaynaklı Retina Nakli: Hastanın kendi iPS hücrelerinden üretilen fotoreseptör tabakalarının nakli üzerine araştırmalar devam etmektedir.
Yapay Retina (Retina Protezi): Son dönem RP için elektriksel stimülasyon cihazları. Argus II ve diğerleri yurtdışında kullanıma sunulmuştur ve Japonya’da suprakoroidal transretinal stimülasyon yöntemiyle klinik deneyler devam etmektedir.
QGen tedavisi Japonya'da da mevcut mu?
A
Voretigen neparvovek (Luxturna) 2023’te Japonya’da onaylanmıştır, ancak yalnızca RPE65 genindeki biallelik patojenik varyantlara sahip retina distrofisi ile sınırlıdır 9)18)19). RPGR mutasyonu dahil diğer gen mutasyonlarına sahip RP için gen tedavisi şu anda klinik deney aşamasındadır 7) ve Japonya’da genel bir tedavi olarak onaylanmamıştır.
QAraştırma aşamasındaki tedavilere nasıl erişebilirim?
A
Klinik deneylere katılım, yalnızca tıp kurumunun etik kurulu tarafından onaylanmış resmi deneylerle sınırlıdır. Sorumlu doktora danışmanın yanı sıra, Ulusal Kanser Merkezi tarafından işletilen Klinik Deney Bilgi Sistemi (jRCT) ve ABD clinicaltrials.gov üzerinde deney bilgileri aranabilir.
Baz-Redón N, Sánchez-Bellver L, Fernández-Cancio M, Rovira-Amigo S, Burgoyne T, Ranjit R, Aquino V, Toro-Barrios N, et al. Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene. Cells. 2024;13(6). doi:10.3390/cells13060524. PMID:38534367; PMCID:PMC10968961.
Holanda IP, et al. Syndromic Retinitis Pigmentosa: A 15-Patient Study Exploring Clinical and Genetic Features. Genes. 2024;15(4):516.
Wawrocka A, Walczak-Sztulpa J, Kuszel L, Niedziela-Schwartz Z, Skorczyk-Werner A, Bernardczyk-Meller J, Krawczynski MR. Coexistence of Retinitis Pigmentosa and Ataxia in Patients with PHARC, PCARP, and Oliver-McFarlane Syndromes. International journal of molecular sciences. 2024;25(11). doi:10.3390/ijms25115759. PMID:38891946; PMCID:PMC11172263.
Lujia Zhang, Ya Li, Litao Qin, Yu Wu, Bo Lei. Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population. Genes. 2021;12(4):537. doi:10.3390/genes12040537.
Daiki Sakai, Seiji Takagi, Yasuhiko Hirami, Makoto Nakamura, Yasuo Kurimoto. Use of ellipsoid zone width for predicting visual prognosis after cataract surgery in patients with retinitis pigmentosa. Eye. 2022;37(1):42-47. doi:10.1038/s41433-021-01878-3.
日本眼科学会. 網膜色素変性診療ガイドライン. 日眼会誌. 2016;120:846-861.
Campochiaro PA, Mir TA. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res. 2018;62:24-37. doi:10.1016/j.preteyeres.2017.08.004.
Liu Y, Zong X, Cao W, Zhang W, Zhang N, Yang N. Gene therapy for retinitis pigmentosa: current challenges and new progress. Biomolecules. 2024;14(8):903. PMID: 39199291. PMCID: PMC11352491. doi:10.3390/biom14080903.
Napoli D, Di Marco B, Salamone G, Orsini N, Mazziotti R, Strettoi E. Keeping the lights on: a new role for an old drug to support cone survival in Retinitis Pigmentosa. Progress in retinal and eye research. 2025;109:101403. doi:10.1016/j.preteyeres.2025.101403. PMID:40998215.
Koyanagi Y, Akiyama M, Nishiguchi KM, Momozawa Y, Kamatani Y, Takata S, et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. Journal of medical genetics. 2019;56(10):662-670. doi:10.1136/jmedgenet-2018-105691. PMID:31213501.
Numa S, Oishi A, Higasa K, Oishi M, Miyata M, Hasegawa T, Ikeda HO, Otsuka Y, Matsuda F, Tsujikawa A.. EYS is a major gene involved in retinitis pigmentosa in Japan: genetic landscapes revealed by stepwise genetic screening. Sci Rep. 2020;10(1):20770. doi:10.1038/s41598-020-77558-1. PMID:33247286; PMCID:PMC7695703.
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, Willett W.. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. doi:10.1001/archopht.1993.01090060049022. PMID:8512476.
Nakazawa M, Suzuki Y, Ito T, Metoki T, Kudo T, Ohguro H. Long-term effects of nilvadipine against progression of the central visual field defect in retinitis pigmentosa: an extended study. BioMed research international. 2013;2013:585729. doi:10.1155/2013/585729. PMID:24319686; PMCID:PMC3844269.
Yamamoto S, Sugawara T, Murakami A, Nakazawa M, Nao-I N, Machida S, et al. Topical isopropyl unoprostone for retinitis pigmentosa: microperimetric results of the phase 2 clinical study. Ophthalmology and therapy. 2012;1(1):5. doi:10.1007/s40123-012-0005-9. PMID:25135585; PMCID:PMC4108136.
Campochiaro PA, Iftikhar M, Hafiz G, Akhlaq A, Tsai G, Wehling D, et al. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. The Journal of clinical investigation. 2020;130(3):1527-1541. doi:10.1172/JCI132990. PMID:31805012; PMCID:PMC7269599.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.