La retinite pigmentosa (RP) è un termine generico per un gruppo di malattie ereditarie caratterizzate da una degenerazione progressiva e diffusa dei fotorecettori (bastoncelli e coni) e dell’epitelio pigmentato retinico (EPR). La degenerazione dei bastoncelli precede quella dei coni; questa viene chiamata distrofia dei bastoncelli e dei coni, e la RP è considerata sinonimo. Non è una singola malattia ma un gruppo di malattie che coinvolgono oltre 100 geni.
La prevalenza è di 1 persona su 4.000-8.000 e il numero totale di pazienti in Giappone supera almeno 30.000 (20.687 beneficiari della malattia rara designata nel 2023) 9). Per quanto riguarda il deficit visivo, è la seconda causa di deficit visivo (13,0%) tra i nuovi titolari di certificato di disabilità visiva di età pari o superiore a 18 anni (dopo il glaucoma, 40,7% nel 2019) e la prima causa di cecità congenita 9). In Giappone è riconosciuta come malattia rara designata ai sensi della legge sulle malattie rare (dal 1° gennaio 2015) 9) e dà diritto a un contributo per le spese mediche.
La RP esiste anche come RP sindromica associata ad altre malattie sistemiche, e la ciliopatia è classificata come concetto sovraordinato come segue 9)2).
Ciliopatia (ciliopathy) :
Sindrome di Usher (tipo 1/2/3) : RP + sordità; malattia rara designata (AR). Il tipo 1 presenta sordità grave e disfunzione vestibolare fin dalla prima infanzia.
Mucopolisaccaridosi (Hurler, Hunter) : con opacità del fondo oculare
Malattia di Refsum (forma adulta e infantile) : malattia perossisomiale; atassia cerebellare, polineuropatia (AR)
Sindrome di Bassen-Kornzweig : alterazione del metabolismo lipidico
Malattia mitocondriale :
Sindrome di Kearns-Sayre : oftalmoplegia esterna progressiva bilaterale, ptosi, disturbo della conduzione cardiaca
Distrofia muscolare:
Distrofia miotonica: può essere associata a RP
Inoltre, è importante la diagnosi differenziale con varie sindromi come PHARC (polineuropatia, ipoacusia, atassia, RP, cataratta), PCARP e sindrome di Oliver-McFarlane3).
QLa retinite pigmentosa è ereditaria?
A
La RP è una malattia genetica, ma non viene necessariamente trasmessa a tutti i figli. Il rischio di trasmissione ai figli varia a seconda della modalità di ereditarietà. Nella forma autosomica dominante (AD) il rischio per i figli è del 50%, mentre nelle forme autosomica recessiva (AR) o legata all’X (XL) il rischio varia in base alla modalità. Nei casi sporadici (48-63% del totale) il rischio per la generazione successiva è spesso relativamente basso9). Si raccomanda il ricorso alla consulenza genetica.
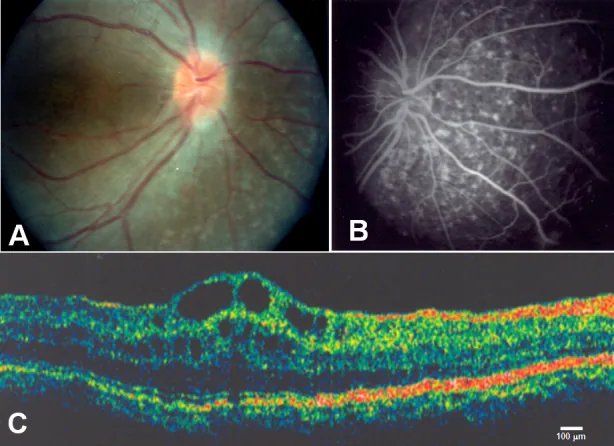
Zenteno JC, et al. Compound heterozygosity for a novel and a recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis. 2009. Figure 1. PMCID: PMC2742641. License: CC BY.
A: drusen del disco ottico e atrofia estesa dell’epitelio pigmentato retinico; B: iperfluorescenza coroidale corrispondente all’atrofia dell’epitelio pigmentato retinico; C: edema maculare cistoide e separazione degli strati retinici interni a livello della fovea. Corrisponde all’edema maculare cistoide trattato nella sezione «2. Principali sintomi e reperti clinici».
I sintomi della RP cambiano in base allo stadio della malattia. Poiché i bastoncelli degenerano per primi, il sintomo più precoce è la cecità notturna.
Cecità notturna: riduzione dell’acuità visiva e difficoltà a vedere al buio. Compare molto precocemente perché i bastoncelli degenerano per primi9); viene avvertita tra i 10 e i 20 anni come difficoltà a vedere in condizioni di scarsa illuminazione. All’inizio la funzione visiva diurna è spesso normale.
Restringimento del campo visivo: il campo visivo si restringe gradualmente dalla periferia verso il centro. Progredisce da uno scotoma anulare a un restringimento concentrico (visione a tunnel)9).
Riduzione dell’acuità visiva: quando alla degenerazione dei bastoncelli segue quella dei coni, anche la visione centrale si riduce. In presenza di edema maculare cistoide (CME), può verificarsi una riduzione moderata dell’acuità visiva relativamente precoce. In alcuni casi, l’acuità visiva centrale può essere preservata fino allo stadio terminale.
Fotofobia (cecità diurna): ipersensibilità alla luce. Manifestazione di disfunzione dei coni. Aumenta con il progredire della degenerazione dei coni. Importante differenziare dalla diffusione della luce dovuta a cataratta.
Fotopsie: possono verificarsi a causa della degenerazione e della perdita dei fotorecettori.
Allucinazioni visive (sindrome di Charles Bonnet) : Nei pazienti con grave calo della vista, fenomeno per cui vedono paesaggi o persone che non esistono realmente. Non è un’esperienza patologica, ma un fenomeno dovuto all’iperattività della corteccia visiva 9)
Di seguito è riportata una panoramica della progressione dei sintomi per stadio della malattia.
Stadio
Sintomi principali
Periodo indicativo
Iniziale
Cecità notturna
10-20 anni
Intermedio
Restringimento del campo visivo (scotoma anulare → concentrico)
30-40 anni
Avanzato
Calo della vista, anomalie della visione dei colori, fotofobia
50 anni e oltre
Reperti clinici e classificazione dei tipi di malattia
Depositi pigmentari a spicola ossea : pigmentazione caratteristica (bone spicule pattern) che compare dalla media periferia alla periferia.
Restringimento delle arterie retiniche : secondario alla degenerazione dei fotorecettori.
Pallore ceroso della papilla ottica : riflette la degenerazione del nervo ottico.
Classificazione dei tipi : si distinguono forme tipiche e atipiche9).
RP tipica (distrofia dei bastoncelli e dei coni) : i bastoncelli vengono danneggiati per primi, poi i coni.
Distrofia dei bastoncelli (sottotipo) : i coni non vengono danneggiati fino allo stadio terminale; l’acuità visiva centrale è preservata anche in caso di grave restringimento concentrico del campo visivo.
RP atipica9) :
RP senza pigmento : assenza di depositi pigmentari.
RP unilaterale : presente in un solo occhio o con marcata asimmetria.
RP settoriale : limitata a 1-2 quadranti della retina; progressione lenta, buona prognosi.
RP centrale/paracentrale : lesioni retiniche e anomalie del campo visivo iniziano centralmente.
Retinopatia a punti bianchi : lesioni puntiformi bianco-giallastre nella retina.
Nei bambini spesso mancano i segni tipici e l’ERG è la chiave per la diagnosi.
Cataratta sottocapsulare posteriore (CSP) : si verifica in circa il 50% dei casi. Caratterizzata da riduzione della vista in condizioni di luce intensa. Una larghezza della zona ellissoidale (EZ) ≥ 600 μm predice una buona acuità visiva postoperatoria (AUC 0,97) 5).
Edema maculare cistoide (EMC) : si verifica nel 10-50% dei casi ed è la principale causa di riduzione della visione centrale 9).
Glaucoma ad angolo chiuso : sono state riportate crisi in circa l’1% dei casi; può verificarsi anche sublussazione del cristallino a causa della fragilità delle zonule di Zinn 9).
Foro maculare / schisi foveale : relativamente raro, ma può richiedere vitrectomia9).
QLa chirurgia della cataratta migliora la vista?
A
Per la chirurgia della cataratta sottocapsulare posteriore associata a RP, se la larghezza della zona ellissoidale (EZ) all’OCT preoperatorio è ≥ 600 μm, ci si può aspettare una buona acuità visiva postoperatoria (AUC 0,97) 5). La larghezza dell’EZ è un biomarcatore utile per prevedere la funzione visiva preoperatoria. Tuttavia, le zonule di Zinn sono spesso fragili, richiedendo attenzione alla contrazione capsulare anteriore e alla lussazione della IOL. Per la prevenzione dell’EMC postoperatorio (10-14%), si raccomanda un uso prolungato rispetto al normale di colliri steroidei e FANS 9).
La RP è un gruppo di malattie geneticamente eterogenee causate da mutazioni in oltre 100 geni 9). I principali geni causali nei giapponesi sono presentati per modalità di trasmissione.
Confronto dei principali geni causali di seguito.
Gene
Ereditarietà
Frequenza/caratteristiche nei giapponesi
EYS
AR
30-50% dei casi con gene identificato (più frequente nella forma AR) 12)13)
USH2A
AR
Secondo più frequente nella forma AR (4-9%); gene principale della sindrome di Usher12)
RHO
AD
Più frequente nella forma AD 6)
RPGR
X-linked
Circa 70-75% nella forma XL 6)
REEP6
AR
Uno dei geni causali della forma AR 4)
Le caratteristiche di ciascun gene sono integrate di seguito.
EYS (Eyes Shut Homolog) : Gene causale più frequente della RP di forma AR nei giapponesi (30-50% dei casi identificati) 12)13). In Occidente non è così frequente, riflettendo un background genetico specifico dei giapponesi.
USH2A : Gene principale della sindrome di Usher (RP + sordità), secondo più frequente nella RP di forma AR nei giapponesi dopo EYS (4-9%) 12)
RHO (Rodopsina) : gene più frequentemente responsabile della RP di tipo AD6). Codifica per la proteina fotorecettrice dei bastoncelli.
RPGR (Regolatore della GTPasi della Retinite Pigmentosa) : gene principale responsabile della RP di tipo XL6). Sono stati riportati casi di disfunzione ciliare primaria (PCD) in pazienti di sesso maschile con mutazioni di RPGR1).
REEP6 (Proteina 6 che migliora l’espressione del recettore) : uno dei geni responsabili della RP di tipo AR4).
Il tasso di rilevamento dei geni causali tramite test genetici varia a seconda della modalità di trasmissione. È stimato al 35-60% per il tipo AD, al 30-50% per il tipo AR e i casi sporadici, e al 16-36% per il tipo XL6).
Inoltre, nella RP sindromica, inclusa la sindrome di Joubert, la sindrome di Bardet-Biedl, ecc., sono frequenti mutazioni nei geni correlati alle ciglia e possono essere associate a complicanze sistemiche (malattie renali, polidattilia, obesità, ecc.)2). È importante anche la diagnosi differenziale con varie sindromi come l’atassia di Friedreich (PHARC), PCARP, sindrome di Oliver-McFarlane, ecc.3).
QÈ opportuno sottoporsi a un test genetico?
A
La diagnosi genetica è importante per la diagnosi definitiva, la consulenza genetica e la determinazione dell’idoneità alla terapia genica. Il sistema PrismGuide IRD Panel (analisi completa delle sequenze esoniche di 82 geni patogeni della IRD) è stato approvato per la copertura assicurativa nel 2023, ma a giugno 2025 sono idonei solo i pazienti giovani con sospetta IRD correlata a RPE659). Si raccomanda di eseguirla in combinazione con la consulenza genetica. È possibile ricevere una consulenza genetica senza aver effettuato una diagnosi genetica.
La diagnosi di RP si basa su una combinazione di reperti clinici, esami elettrofisiologici, imaging e test genetici.
I criteri diagnostici (criteri di certificazione)9) includono i seguenti elementi:
A. Sintomi (almeno uno) :
Sintomi soggettivi progressivi
Almeno uno tra: cecità notturna, restringimento del campo visivo, riduzione dell’acuità visiva, fotofobia
B. Reperti di esame (almeno due) :
(1) Reperti del fondo oculare: restringimento dei vasi retinici, aspetto ruvido della retina, depositi pigmentari a forma di corpuscoli ossei, punti bianchi multipli, atrofia del nervo ottico, degenerazione maculare
Anello iperfluorescente anomalo (AF ring) come indicatore di stadio 6)
Esame del campo visivo
Valutazione della progressione
Perimetro di Goldmann; anche il programma HFA 10-2 è utile 9)10)
Esame della visione dei colori
Valutazione della funzione dei coni
Anomalia blu-giallo acquisita frequente; test Panel D-15 e 100 Hue 9)
Esame di adattamento al buio
Valutazione della funzione dei bastoncelli
Punto di flesso di Kohlrausch non rilevato 10)
Test genetico NGS
Diagnosi genetica
Pannello PrismGuide IRD (82 geni) 9)
I dettagli di ciascun esame sono riportati di seguito.
Elettroretinografia (ERG) : Essenziale per la diagnosi definitiva 6)9). La risposta dei bastoncelli (ERG scotopico) è ridotta fin dall’inizio e, con la progressione, anche la risposta dei coni (ERG fotopico) si riduce. L’ERG full-field è la base. Spesso al momento della visita è già non registrabile.
OCT (tomografia a coerenza ottica) : Valuta la larghezza della zona ellissoidale (EZ) e il pattern di scomparsa. La larghezza dell’EZ è utile come biomarcatore quantitativo della funzione visiva e della prognosi, ed è utilizzata anche per decidere l’indicazione all’intervento di cataratta5). Fin dall’inizio della malattia si osservano assottigliamento dello strato nucleare esterno e scomparsa dell’EZ.
Autofluorescenza del fondo (FAF) : Un anello iperfluorescente anomalo (anello autofluorescente; AF ring) appare intorno alla macula, indicando la retina funzionante residua e fungendo da indicatore di progressione della malattia 6).
Esame del campo visivo : La perimetria dinamica con il perimetro di Goldmann è lo standard. Con la progressione si osserva uno scotoma anulare, seguito da un restringimento concentrico del campo visivo 10). Il programma 10-2 del perimetro di Humphrey (HFA) è utile per valutare la funzione residua dei coni centrali 9).
Esame della visione dei colori : Si osserva frequentemente un deficit acquisito blu-giallo. La valutazione viene effettuata con il test Panel D-15 e il test 100 Hue 9).
Esame di adattamento al buio : Il punto di flesso di Kohlrausch (punto di passaggio tra bastoncelli e coni) non viene rilevato 10).
Sequenziamento di nuova generazione (NGS) : Il sistema di pannelli PrismGuide IRD consente di analizzare in modo completo le sequenze esoniche di 82 geni patogeni 9). È anche indispensabile per determinare l’indicazione alla terapia genica.
Attualmente non esiste una cura per la RP6)9). Il trattamento si concentra sul mantenimento della funzione visiva, sulla gestione delle complicanze e sul supporto alla vita sociale.
Protezione dei fotorecettori
Vitamina A (15.000 UI/giorno) : La somministrazione orale ritarda il deterioramento dell’ERG di alcuni punti percentuali secondo alcuni rapporti14). Nessun effetto di miglioramento dell’acuità visiva o del campo visivo. Monitoraggio della funzionalità epatica necessario per uso a lungo termine. Controindicato in gravidanza per teratogenicità. Può favorire la progressione nelle mutazioni ABCA414). La vitamina E può accelerare la progressione, quindi è necessaria cautela14).
Collirio di unoprostone : È stata osservata una tendenza al miglioramento dose-dipendente della sensibilità, ma l’endpoint primario dello studio di fase 2 (sensibilità retinica centrale a 2 gradi) non ha mostrato differenze significative16).
Nilvadipina (antagonista del calcio) : Un rapporto a lungo termine ha mostrato un rallentamento della progressione dei difetti del campo visivo15). Si tratta di un rapporto monocentrico con un numero limitato di casi; non sono stati condotti studi multicentrici di follow-up.
N-acetilcisteina (NAC) : Soppressione dello stress ossidativo. Uno studio di fase I ha riportato un miglioramento dell’acuità visiva17) e nel 2025 è in corso uno studio di fase III.
DHA e luteina : Proteggono i fotorecettori maculari dallo stress ossidativo. Non è stato osservato alcun effetto additivo del DHA alla vitamina A.
Elenio (Adaptinol) : Approvato per il miglioramento temporaneo del campo visivo e dell’adattamento al buio nella RP. Non è stata condotta una valutazione dell’efficacia secondo gli standard medici moderni.
Occhiali schermanti : Riduce lo stress ossidativo da UV e luce intensa. Si raccomanda l’uso quotidiano.
La prima linea è un inibitore dell’anidrasi carbonica (CAI). Utilizzo di collirio di dorzolamide (Trusopt) o acetazolamide (Diamox) per via orale. Un miglioramento dello spessore maculare centrale si ottiene in circa il 40% dei casi. Si osserva una recidiva in circa il 30% dei casi9).
I farmaci anti-VEGF non sono raccomandati nella RP-CME poiché la produzione di VEGF è ridotta9).
Si noti che nessuno di questi trattamenti è approvato per la RP-CME e sono usi off-label.
Chirurgia della cataratta: eseguita in caso di cataratta sottocapsulare posteriore. Una larghezza della zona ellissoidale (EZ) ≥ 600 μm all’OCT preoperatorio è un fattore predittivo di buona acuità visiva postoperatoria 5). In caso di fragilità delle zonule di Zinn, considerare l’uso di un anello di tensione capsulare. Per prevenire l’edema maculare cistoide postoperatorio (10-14%), utilizzare colliri steroidei e FANS per un periodo più lungo del solito 9).
Glaucoma ad angolo chiuso: il rischio di glaucoma primario ad angolo chiuso è elevato nei pazienti con RP. La camera anteriore diventa progressivamente più superficiale; eseguire profilatticamente iridotomia laser o chirurgia della cataratta9).
Membrana epiretinica (GL2026 CQ4): vitrectomia. Nei casi con linea EZ continua ci si può aspettare un miglioramento dell’acuità visiva. Nei casi con linea EZ discontinua il recupero è limitato. Sono stati riportati casi di atrofia maculare grave a lungo termine; è opportuna una valutazione in un centro specializzato 9).
Foro maculare: la vitrectomia è l’unico trattamento curativo. I risultati postoperatori sono poco studiati 9).
Supporto e riabilitazione
Cura della bassa visione: ipovisione → lenti d’ingrandimento, ingranditori elettronici, tablet; fotofobia → occhiali schermanti; restringimento del campo visivo → bastone bianco; visione da lontano → monoculare; occhiali per la visione notturna. È importante un supporto individualizzato in base al campo visivo e all’acuità. Si raccomanda l’uso di Smart Site (presentazione dei servizi di consulenza per la bassa visione in ciascuna regione).
Consulenza genetica: fornita da un genetista clinico e un consulente genetico certificato. Le consultazioni riguardano spesso la stima del rischio di ricorrenza, istruzione, lavoro, matrimonio e procreazione. È possibile ricevere una consulenza genetica senza aver effettuato una diagnosi genetica.
Sistema per malattie rare: come malattia rara designata, è disponibile il sistema di assistenza per le spese mediche 9). Considerare anche l’ottenimento del certificato di invalidità e dell’assistenza medica per l’autonomia.
Voretigene neparvovec (Luxturna): farmaco di terapia genica somministrabile a pazienti con varianti patogene bialleliche del gene RPE65 e sufficienti cellule retiniche vitali. Approvato in Giappone nel 2023 9). Nello studio di fase III statunitense (301) sono stati arruolati 31 pazienti; l’analisi mITT (intervento 20, controllo 9) ha mostrato un miglioramento significativo di MLMT e FST con luce bianca rispetto al gruppo di controllo 18). Nello studio di fase III nazionale (A11301) è stato confermato un aumento della sensibilità FST e un’espansione del campo visivo in 4 pazienti giapponesi 19). L’effetto di miglioramento dell’acuità visiva è scarso e come complicanza a lungo termine è stata riportata atrofia retinocoroideale in oltre il 20% dei pazienti 9).
Eseguire ogni 6 mesi-1 anno: acuità visiva, lampada a fessura, fundus, campo visivo Humphrey (HFA 10-2), OCT9).
QQuali farmaci vengono utilizzati per trattare l'edema maculare?
A
Il trattamento di prima linea per l’EMC-RP sono gli inibitori dell’anidrasi carbonica (IAC), somministrati come collirio a base di dorzolamide o acetazolamide per via orale 9). Un miglioramento dello spessore maculare centrale si ottiene in circa il 40% dei casi, ma in circa il 30% si verifica una recidiva. In caso di mancato miglioramento con gli IAC, si possono prendere in considerazione iniezioni intravitreali di triamcinolone acetonide o un impianto intravitreale di desametasone (Ozurdex). I farmaci anti-VEGF non sono raccomandati nell’EMC-RP. Si noti che tutti questi usi sono off-label.
La via finale comune della morte dei fotorecettori nella RP è l’apoptosi. Sebbene i tipi di mutazioni genetiche siano diversi, alla fine convergono in una via comune di morte cellulare.
Degenerazione secondaria dei coni dopo i bastoncelli
Nella RP, il pattern tipico è la degenerazione e la scomparsa prima dei bastoncelli, seguita dalla degenerazione secondaria dei coni 7). Si ritiene che i coni dipendano da un fattore di sopravvivenza prodotto dai bastoncelli (RdCVF: Rod-derived Cone Viability Factor); pertanto, dopo la perdita dei bastoncelli, anche i coni perdono la loro funzione 7)11).
La retina è uno dei tessuti metabolicamente più attivi; converte l’80-90% del glucosio in lattato attraverso la glicolisi aerobica (effetto Warburg). I coni sono più vulnerabili allo stress metabolico rispetto ai bastoncelli, e questa vulnerabilità metabolica contribuisce anche alla degenerazione secondaria dei coni 11).
Anche l’infiammazione è riconosciuta come un fattore principale nella progressione della RP; l’attivazione della microglia e l’infiltrazione di macrofagi aggravano il danno retinico 11). Lo stress ossidativo agisce anche come driver biologico della degenerazione secondaria dei coni.
I meccanismi di degenerazione differiscono a seconda del gene.
Mutazioni RHO: La rodopsina mal ripiegata induce stress del reticolo endoplasmatico → UPR (risposta alle proteine non ripiegate) → apoptosi11)
Mutazione REEP6 : REEP6 codifica per una proteina coinvolta nel mantenimento della morfologia del RE. Le mutazioni patogene portano alla formazione di corpi di inclusione del RE nel segmento esterno dei bastoncelli, con conseguente degenerazione dei fotorecettori4)
Mutazione RPGR : RPGR è coinvolto nella struttura assonemale dei cigli primari; le mutazioni compromettono il trasporto di sostanze verso il segmento esterno dei fotorecettori1)
7. Ricerche recenti e prospettive future (rapporti in fase di ricerca)
La terapia genica è l’approccio più promettente per il trattamento delle malattie retiniche ereditarie8).
Luxturna (voretigene neparvovec) : Farmaco di terapia genica somministrabile a pazienti con varianti patogene bialleliche del gene RPE65. Nello studio di fase III statunitense (studio 301), sono stati arruolati 31 pazienti; l’analisi mITT (20 intervento, 9 controllo) ha mostrato un miglioramento significativo del MLMT e del FST con luce bianca rispetto al gruppo di controllo18). Nello studio di fase III nazionale (studio A11301), è stato confermato un aumento della sensibilità FST e un’espansione del campo visivo in 4 pazienti giapponesi19). Approvato in Giappone nel 2023, funge da ponte tra il trattamento standard e la terapia sperimentale.
Terapia genica per RPGR : La terapia genica mediata da AAV per la RP legata all’X dovuta a mutazioni RPGR è progredita fino agli studi clinici di fase I/II/III8).
CRISPR/Cas9 : Sono in corso ricerche per la correzione diretta di mutazioni patogene o l’inattivazione di mutazioni dominanti negative8).
RdCVF (fattore di sopravvivenza dei coni derivato dai bastoncelli) e terapia di protezione dei coni
RdCVF è una proteina secreta dai bastoncelli che mantiene la sopravvivenza dei coni7)11). Sono in corso studi clinici sulla terapia di protezione dei coni utilizzando RdCVF, considerata una strategia terapeutica indipendente per preservare la funzione dei coni dopo la degenerazione dei bastoncelli.
La NAC è un farmaco che sopprime lo stress ossidativo; uno studio di fase I ha riportato un miglioramento dell’acuità visiva17). Nel 2025 è in corso uno studio di fase III.
Potenziale di riposizionamento dei glucocorticoidi (desametasone)
Recenti studi in vivo (modello murino rd10) hanno dimostrato che il desametasone intravitreale protegge i fotorecettori a cono e l’epitelio pigmentato retinico11). I glucocorticoidi hanno un forte potenziale di riposizionamento come terapia indipendente dalla mutazione. Tuttavia, le attuali evidenze sono limitate a modelli animali e per l’applicazione clinica nell’uomo è necessaria un’ulteriore validazione.
Trapianto retinico derivato da cellule iPS e retina artificiale
Trapianto retinico derivato da cellule iPS: La ricerca sul trapianto di foglietti di fotorecettori prodotti dalle cellule iPS del paziente è in corso.
Retina artificiale (protesi retinica): Dispositivo di stimolazione elettrica per RP terminale. Argus II e altri sono commercializzati all’estero, e in Giappone sono in corso studi clinici sulla stimolazione transretinica sopracoroideale.
QLa terapia genica è disponibile in Giappone?
A
Il voretigene neparvovec (Luxturna) è stato approvato in Giappone nel 2023, ma solo per le distrofie retiniche con varianti patogene bialleliche del gene RPE65 9)18)19). La terapia genica per la RP con altre mutazioni geniche, incluse le mutazioni RPGR, è ancora in fase di sperimentazione clinica 7) e non è approvata come trattamento comune in Giappone.
QCome accedere ai trattamenti sperimentali?
A
La partecipazione a studi clinici è limitata a studi ufficiali approvati dal comitato etico dell’istituto medico. Oltre a consultare il medico curante, è possibile cercare informazioni sugli studi sul sito di informazioni sugli studi clinici del Centro nazionale per il cancro (jRCT) o sul sito americano clinicaltrials.gov.
Baz-Redón N, Sánchez-Bellver L, Fernández-Cancio M, Rovira-Amigo S, Burgoyne T, Ranjit R, Aquino V, Toro-Barrios N, et al. Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene. Cells. 2024;13(6). doi:10.3390/cells13060524. PMID:38534367; PMCID:PMC10968961.
Holanda IP, et al. Syndromic Retinitis Pigmentosa: A 15-Patient Study Exploring Clinical and Genetic Features. Genes. 2024;15(4):516.
Wawrocka A, Walczak-Sztulpa J, Kuszel L, Niedziela-Schwartz Z, Skorczyk-Werner A, Bernardczyk-Meller J, Krawczynski MR. Coexistence of Retinitis Pigmentosa and Ataxia in Patients with PHARC, PCARP, and Oliver-McFarlane Syndromes. International journal of molecular sciences. 2024;25(11). doi:10.3390/ijms25115759. PMID:38891946; PMCID:PMC11172263.
Lujia Zhang, Ya Li, Litao Qin, Yu Wu, Bo Lei. Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population. Genes. 2021;12(4):537. doi:10.3390/genes12040537.
Daiki Sakai, Seiji Takagi, Yasuhiko Hirami, Makoto Nakamura, Yasuo Kurimoto. Use of ellipsoid zone width for predicting visual prognosis after cataract surgery in patients with retinitis pigmentosa. Eye. 2022;37(1):42-47. doi:10.1038/s41433-021-01878-3.
日本眼科学会. 網膜色素変性診療ガイドライン. 日眼会誌. 2016;120:846-861.
Campochiaro PA, Mir TA. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res. 2018;62:24-37. doi:10.1016/j.preteyeres.2017.08.004.
Liu Y, Zong X, Cao W, Zhang W, Zhang N, Yang N. Gene therapy for retinitis pigmentosa: current challenges and new progress. Biomolecules. 2024;14(8):903. PMID: 39199291. PMCID: PMC11352491. doi:10.3390/biom14080903.
Napoli D, Di Marco B, Salamone G, Orsini N, Mazziotti R, Strettoi E. Keeping the lights on: a new role for an old drug to support cone survival in Retinitis Pigmentosa. Progress in retinal and eye research. 2025;109:101403. doi:10.1016/j.preteyeres.2025.101403. PMID:40998215.
Koyanagi Y, Akiyama M, Nishiguchi KM, Momozawa Y, Kamatani Y, Takata S, et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. Journal of medical genetics. 2019;56(10):662-670. doi:10.1136/jmedgenet-2018-105691. PMID:31213501.
Numa S, Oishi A, Higasa K, Oishi M, Miyata M, Hasegawa T, Ikeda HO, Otsuka Y, Matsuda F, Tsujikawa A.. EYS is a major gene involved in retinitis pigmentosa in Japan: genetic landscapes revealed by stepwise genetic screening. Sci Rep. 2020;10(1):20770. doi:10.1038/s41598-020-77558-1. PMID:33247286; PMCID:PMC7695703.
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, Willett W.. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. doi:10.1001/archopht.1993.01090060049022. PMID:8512476.
Nakazawa M, Suzuki Y, Ito T, Metoki T, Kudo T, Ohguro H. Long-term effects of nilvadipine against progression of the central visual field defect in retinitis pigmentosa: an extended study. BioMed research international. 2013;2013:585729. doi:10.1155/2013/585729. PMID:24319686; PMCID:PMC3844269.
Yamamoto S, Sugawara T, Murakami A, Nakazawa M, Nao-I N, Machida S, et al. Topical isopropyl unoprostone for retinitis pigmentosa: microperimetric results of the phase 2 clinical study. Ophthalmology and therapy. 2012;1(1):5. doi:10.1007/s40123-012-0005-9. PMID:25135585; PMCID:PMC4108136.
Campochiaro PA, Iftikhar M, Hafiz G, Akhlaq A, Tsai G, Wehling D, et al. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. The Journal of clinical investigation. 2020;130(3):1527-1541. doi:10.1172/JCI132990. PMID:31805012; PMCID:PMC7269599.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.