La retinitis pigmentosa (RP) es un término general para un grupo de enfermedades hereditarias caracterizadas por una degeneración progresiva y generalizada que afecta principalmente a los fotorreceptores (bastones y conos) y al epitelio pigmentario de la retina (EPR). Cuando la degeneración de los bastones precede a la de los conos, se denomina distrofia de bastones y conos, y la RP se entiende como sinónimo. No es una enfermedad única, sino un grupo de trastornos que involucran más de 100 genes.
La prevalencia es de 1 en 4.000 a 8.000, y el número total de pacientes en Japón supera al menos los 30.000 (20.687 beneficiarios de la prestación por enfermedad intratable designada en el año fiscal 2023) 9). En cuanto a la discapacidad visual, la RP es la segunda causa (13,0%) entre los nuevos titulares de certificados de discapacidad física mayores de 18 años (después del glaucoma con 40,7% en el año fiscal 2019) y la primera causa de ceguera congénita 9). En Japón, ha sido designada como enfermedad intratable según la Ley de Enfermedades Intratables (desde el 1 de enero de 2015) 9) y es elegible para subsidios de gastos médicos.
La RP también incluye RP sindrómico acompañado de otras enfermedades sistémicas, y se clasifica de la siguiente manera con la ciliopatía como concepto superior 9)2).
Ciliopatía:
Síndrome de Usher (tipo 1/2/3): RP + pérdida auditiva; enfermedad intratable designada (AR). El tipo 1 presenta pérdida auditiva severa y disfunción vestibular desde la infancia temprana.
Síndrome de Bardet-Biedl: obesidad, retraso del desarrollo psicomotor, polidactilia, hipoplasia genital (AR)
Síndrome de Senior-Løken: RP + nefritis juvenil (AR)
Síndrome de Alström: RP + obesidad, pérdida auditiva, diabetes (AR)
Mucopolisacaridosis (Hurler, Hunter): acompañada de opacidad del fondo de ojo
Enfermedad de Refsum (tipo adulto, tipo infantil): enfermedad peroxisomal; ataxia cerebelosa, polineuropatía (AR)
Síndrome de Bassen-Kornzweig: trastorno del metabolismo lipídico
Enfermedad mitocondrial:
Síndrome de Kearns-Sayre: oftalmoplejía externa progresiva bilateral, ptosis palpebral, trastorno de la conducción cardíaca
Distrofia muscular:
Distrofia miotónica: Puede asociarse con RP
Además, es importante el diagnóstico diferencial con diversos síndromes como PHARC (polineuropatía, hipoacusia, ataxia, RP, catarata), PCARP y síndrome de Oliver-McFarlane 3).
Q¿La retinitis pigmentosa es hereditaria?
A
La RP es una enfermedad hereditaria, pero no necesariamente se hereda en todos los casos. El riesgo de herencia para los hijos varía según el patrón de herencia. En el tipo AD, existe un 50% de probabilidad de herencia para los hijos, pero en los tipos AR y XL, el riesgo varía según el patrón de herencia. En los casos esporádicos (48-63% del total), el riesgo de herencia para la siguiente generación suele ser relativamente bajo 9). Se recomienda el uso de asesoramiento genético.
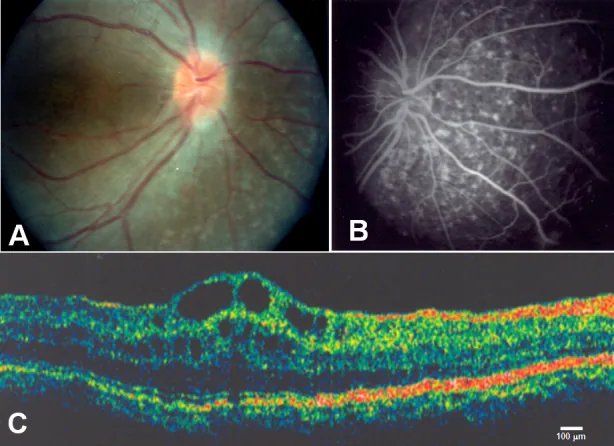
Zenteno JC, et al. Compound heterozygosity for a novel and a recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis. 2009. Figure 1. PMCID: PMC2742641. License: CC BY.
A muestra drusas del disco óptico y atrofia extensa del epitelio pigmentario de la retina, B muestra hiperfluorescencia coroidea correspondiente a la atrofia del epitelio pigmentario de la retina, C muestra edema macular quístico y separación de las capas internas de la retina en la fóvea. Esto corresponde al edema macular quístico tratado en la sección “2. Síntomas principales y hallazgos clínicos”.
Los síntomas de la RP cambian según la etapa de progresión. Dado que los fotorreceptores de bastones degeneran primero, la ceguera nocturna aparece como el síntoma más temprano.
Ceguera nocturna: Disminución de la visión o dificultad para ver en lugares oscuros. Aparece desde la etapa más temprana porque los bastones degeneran primero 9); se nota como dificultad para ver en lugares con poca luz en la segunda y tercera década de la vida. En las etapas iniciales, la función visual diurna suele ser normal.
Estrechamiento del campo visual: Se estrecha gradualmente desde la periferia hacia adentro. Progresa de escotoma anular a estrechamiento concéntrico del campo visual (visión en túnel) 9)
Disminución de la agudeza visual: Cuando la degeneración de los conos progresa después de la de los bastones, la agudeza visual central también disminuye. Cuando se asocia CME, puede ocurrir una pérdida moderada de la agudeza visual relativamente temprano. En algunos casos, la agudeza visual central se conserva hasta la etapa final.
Fotofobia (hemeralopía): Sensibilidad a la luz. Manifestación de disfunción de los conos. Aumenta a medida que progresa la degeneración de los conos. Es importante diferenciarla de la dispersión de la luz debida a cataratas.
Fotopsia: Puede ocurrir con la degeneración y pérdida de fotorreceptores.
Alucinaciones visuales (síndrome de Charles Bonnet): Fenómeno en el que pacientes con pérdida visual avanzada ven paisajes o personas que no existen realmente. No es una experiencia patológica, sino un fenómeno causado por la hiperactividad de la corteza visual 9)
A continuación se muestra la progresión aproximada de los síntomas según la etapa de la enfermedad.
Etapa
Síntomas principales
Edad aproximada
Inicial
Ceguera nocturna
10–20 años
Intermedia
Estrechamiento del campo visual (escotoma anular → concéntrico)
30–40 años
Avanzada
Pérdida de visión, anomalías de la visión cromática, fotofobia
Pigmentación en espículas óseas: Pigmentación característica (patrón en espículas óseas) que aparece desde la periferia media hasta la periferia
Atenuación de las arterias retinianas: Ocurre secundariamente a la degeneración de los fotorreceptores
Palidez cérea del disco óptico: Refleja la degeneración del nervio óptico
Clasificación incluye formas típicas y atípicas9).
RP típica (distrofia de conos y bastones): Los bastones se afectan primero, seguidos más tarde por los conos
Distrofia de bastones (subtipo): Los conos no se afectan hasta la etapa terminal; la visión central se conserva incluso con una constricción concéntrica severa del campo visual
RP atípica9):
Retinitis pigmentosa sine pigmento: No se observa pigmentación
RP unilateral: Afecta solo un ojo, o asimetría significativa entre ambos ojos
RP sectorial: Limitada a 1–2 cuadrantes de la retina; progresión lenta, buen pronóstico
RP central/pericentral: Las lesiones retinianas y las anomalías del campo visual se originan centralmente
Retinitis punctata albescens: Lesiones puntiformes blancas a amarillas en la retina
En niños, los hallazgos típicos suelen estar incompletos, y el ERG es clave para el diagnóstico.
Catarata subcapsular posterior (CSP): Ocurre en aproximadamente el 50% de los casos. Se caracteriza por disminución de la visión en lugares iluminados. Un ancho de la zona elipsoide (EZ) de 600 μm o más predice una buena agudeza visual postoperatoria (AUC 0.97)5).
Edema macular quístico (EMQ): Ocurre en el 10–50% de los casos y es una causa principal de pérdida de la visión central9).
Glaucoma de ángulo cerrado: Se han reportado ataques en aproximadamente el 1% de los casos; también puede ocurrir subluxación del cristalino debido a la debilidad de las zónulas de Zinn9).
Agujero macular / foveosquisis: Relativamente raro, pero puede ser indicación de vitrectomía9).
Q¿Mejora la visión después de la cirugía de cataratas?
A
En la cirugía de cataratas para la catarata subcapsular posterior asociada a RP, se puede esperar una buena agudeza visual postoperatoria si la OCT preoperatoria muestra un ancho de la zona elipsoide (EZ) de 600 μm o más (AUC 0.97)5). El ancho de la EZ es un biomarcador útil para predecir la función visual antes de la cirugía. Sin embargo, las zónulas suelen ser frágiles, por lo que se debe tener precaución ante la contracción capsular anterior y el desplazamiento del LIO. Para la prevención del EMQ postoperatorio (10–14%), se recomienda el uso prolongado de gotas oftálmicas de esteroides y AINEs más allá de lo habitual9).
La RP es un grupo de enfermedades con alta heterogeneidad genética causada por mutaciones en más de 100 genes9). A continuación se muestran los principales genes causantes en pacientes japoneses según el patrón de herencia.
A continuación se muestra una comparación de los principales genes causantes.
Gen
Herencia
Frecuencia/Características en japoneses
EYS
AR
30–50% de los casos con gen causante identificado (más común en tipo AR)12)13)
USH2A
AR
Segundo más frecuente en tipo AR (4–9%); gen principal del síndrome de Usher12)
RHO
AD
Más frecuente en tipo AD6)
RPGR
Ligado al X
Alrededor del 70–75% del tipo XL6)
REEP6
AR
Uno de los genes causantes del tipo AR4)
A continuación se complementan las características de cada gen.
EYS (Eyes Shut Homolog): El gen causante más frecuente de RP tipo AR en japoneses (30–50% de los casos identificados)12)13). No es tan frecuente en poblaciones occidentales, lo que refleja un trasfondo genético específico de los japoneses.
USH2A: Gen principal del síndrome de Usher (RP + pérdida auditiva), y el segundo más frecuente en RP tipo AR en japoneses después de EYS (4–9%)12)
RHO (Rodopsina): El gen causante más común de la RPAD 6). Codifica la proteína fotorreceptora en los bastones.
RPGR (Regulador de GTPasa de Retinitis Pigmentosa): El principal gen causante de la RPXL 6). Se han reportado pacientes varones con mutaciones en RPGR que presentan discinesia ciliar primaria (PCD) 1).
REEP6 (Proteína 6 Potenciadora de la Expresión de Receptores): Uno de los genes causantes de la RPAR 4).
La tasa de detección de genes causantes mediante pruebas genéticas varía según el patrón de herencia. Se reporta entre 35-60% para AD, 30-50% para AR y casos esporádicos, y 16-36% para XL 6).
Además, en la RP sindrómica, que incluye el síndrome de Joubert y el síndrome de Bardet-Biedl, son frecuentes las mutaciones en genes relacionados con los cilios y pueden acompañarse de complicaciones sistémicas (enfermedad renal, polidactilia, obesidad) 2). También es importante el diagnóstico diferencial con diversos síndromes como PHARC, PCARP y el síndrome de Oliver-McFarlane 3).
Q¿Debería realizarme una prueba genética?
A
El diagnóstico genético es importante para el diagnóstico definitivo, el asesoramiento genético y la determinación de la elegibilidad para la terapia génica. El sistema de panel PrismGuide IRD (análisis integral de las secuencias de exones de 82 genes causantes de IRD) fue cubierto por el seguro en 2023, pero a junio de 2025 solo está indicado para pacientes de inicio joven con sospecha de IRD relacionada con RPE65 9). Se recomienda realizarlo en combinación con asesoramiento genético. Se puede recibir asesoramiento genético sin someterse a un diagnóstico genético.
El diagnóstico de la RP se realiza combinando hallazgos clínicos, pruebas electrofisiológicas, pruebas de imagen y pruebas genéticas.
Criterios diagnósticos (criterios de certificación)9) incluyen los siguientes elementos:
A. Síntomas (uno o más de los siguientes):
Síntomas subjetivos progresivos
Uno o más de: ceguera nocturna, estrechamiento del campo visual, disminución de la agudeza visual, fotofobia
B. Hallazgos de pruebas (dos o más de los siguientes):
(1) Hallazgos de fondo de ojo: estrechamiento de los vasos retinianos, aspecto rugoso de la retina, pigmentación en espículas óseas, múltiples puntos blancos, atrofia óptica, degeneración macular
Anillo hiperfluorescente anormal (anillo AF) como indicador de estadio6)
Examen de campo visual
Evaluación de progresión
Perimetría de Goldmann; programa HFA 10-2 también útil9)10)
Examen de visión cromática
Evaluación de función de conos
Defecto adquirido azul-amarillo frecuente; prueba Panel D-15 y 100 Hue9)
Examen de adaptación a la oscuridad
Evaluación de función de bastones
Punto de inflexión de Kohlrausch no detectado10)
Prueba genética NGS
Diagnóstico genético
Panel PrismGuide IRD (82 genes) 9)
Los detalles de cada prueba se muestran a continuación.
Electrorretinografía (ERG): Esencial para el diagnóstico definitivo 6)9). La respuesta de bastones (ERG escotópico) disminuye desde etapas tempranas, y con la progresión también disminuye la respuesta de conos (ERG fotópico). El ERG de campo completo es el estándar. A menudo ya no es registrable en el momento de la consulta.
Tomografía de coherencia óptica (OCT): Evalúa el ancho y el patrón de desaparición de la zona elipsoide (EZ). El ancho de la EZ es útil como biomarcador cuantitativo de la función visual y el pronóstico, y también se utiliza para determinar la indicación de cirugía de cataratas 5). Desde etapas tempranas se observan adelgazamiento de la capa nuclear externa y pérdida de la EZ.
Autofluorescencia de fondo (FAF): Aparece un anillo hiperautofluorescente anormal (anillo AF) alrededor de la mácula, que sirve como indicador de la progresión de la enfermedad y de la retina funcional residual 6).
Prueba de campo visual: La perimetría cinética con el perímetro de Goldmann es el estándar. Con la progresión, se presenta un escotoma anular que lleva a una constricción concéntrica del campo visual 10). El programa 10-2 del analizador de campo Humphrey (HFA) es útil para evaluar la función foveal residual de conos 9).
Prueba de visión cromática: Se observa con frecuencia una deficiencia adquirida azul-amarilla. Se evalúa con la prueba Panel D-15 y la prueba de 100 matices 9).
Prueba de adaptación a la oscuridad: No se detecta el punto de inflexión de Kohlrausch (punto de transición bastón-cono) 10).
Secuenciación de nueva generación (NGS): El sistema de panel PrismGuide IRD puede analizar de forma integral las secuencias de exones de 82 genes causantes 9). También es esencial para determinar la elegibilidad para la terapia génica.
Enfermedades adquiridas: Retinopatía autoinmune (AIR), retinopatía asociada a cáncer (CAR), retinopatía asociada a melanoma (MAR) (inicio relativamente agudo; búsqueda de anticuerpos antirretinianos), degeneración retiniana inducida por fármacos (cloroquina, melanina, etc.), degeneración retiniana traumática, infecciosa (rubéola, sífilis, toxoplasmosis), AZOOR (escotoma de campo visual de inicio agudo; fondo de ojo normal inicialmente).
Actualmente, no existe un tratamiento curativo para la RP 6)9). El tratamiento se centra en mantener la función visual, manejar las complicaciones y apoyar la vida social.
Protección de fotorreceptores
Vitamina A (15,000 UI/día): Se ha informado que la administración oral retrasa el deterioro del ERG en un pequeño porcentaje 14). No mejora la agudeza visual ni el campo visual. Se requiere monitoreo de la función hepática para uso prolongado. Contraindicada durante el embarazo por teratogenicidad. Puede acelerar la progresión en mutaciones ABCA4 14). Tenga en cuenta que la vitamina E puede acelerar la progresión y requiere precaución 14).
Gotas de unoprostona: Se observó una tendencia de mejora en la sensibilidad dependiente de la dosis, pero el criterio principal de valoración (sensibilidad retiniana central de 2 grados) en el ensayo de fase 2 no fue significativo 16).
Nilvadipino (bloqueador de canales de calcio): Informes a largo plazo sugieren una progresión más lenta de los defectos del campo visual 15). Basado en informes de un solo centro con muestras pequeñas; no se ha realizado replicación multicéntrica.
N-acetilcisteína (NAC): Suprime el estrés oxidativo. Un ensayo de fase I informó mejoría en la agudeza visual17); fase III en curso a partir de 2025.
DHA y luteína: Protegen los fotorreceptores maculares del estrés oxidativo. No se ha confirmado un beneficio adicional del DHA cuando se agrega a la vitamina A.
Helenien (Adaptinol): Aprobado para la mejora temporal del campo visual y la adaptación a la oscuridad en la RP. No se ha realizado una evaluación de eficacia según los estándares médicos modernos.
Gafas de filtro de luz: Reducen el estrés oxidativo causado por los rayos UV y la luz brillante. Se recomienda su uso diario.
El tratamiento de primera línea son los inhibidores de la anhidrasa carbónica (CAI). Use gotas de dorzolamida (Trusopt) o acetazolamida (Diamox) por vía oral. La mejora del CMT se logra en aproximadamente el 40%. La recurrencia ocurre en aproximadamente el 30% 9).
Los fármacos anti-VEGF no se recomiendan para el CME-RP porque la producción de VEGF está disminuida 9).
Tenga en cuenta que ninguno de estos está aprobado para RP-CME y se usan fuera de indicación.
Cirugía de cataratas: Se realiza en casos con catarata subcapsular posterior. La OCT preoperatoria que muestra un ancho de EZ ≥600 μm es un predictor de buena agudeza visual postoperatoria 5). En casos con debilidad zonular, considere el uso de un anillo de tensión capsular. Para el CME postoperatorio (10-14%), use gotas oftálmicas de esteroides y AINE durante un período más prolongado de lo habitual 9).
Glaucoma de ángulo cerrado: Los pacientes con RP tienen un alto riesgo de desarrollar glaucoma primario de ángulo cerrado. La cámara anterior se vuelve gradualmente poco profunda; realice iridotomía láser profiláctica o cirugía de cataratas 9).
Membrana epirretiniana (GL2026 CQ4): Vitrectomía. Se puede esperar mejoría visual en casos con línea EZ continua. En casos con línea EZ discontinua, la recuperación es limitada. Se ha reportado atrofia macular grave a largo plazo postoperatoriamente; se recomienda evaluación en un centro especializado 9).
Agujero macular: La vitrectomía es el único tratamiento curativo. Los resultados postoperatorios han sido estudiados de forma limitada 9).
Apoyo y Rehabilitación
Cuidado de baja visión: Baja visión → lupas, magnificadores de video, tabletas; fotofobia → gafas tintadas; estrechamiento del campo visual → bastón blanco; visión de lejos → monocular; gafas de ayuda para visión nocturna. Es importante el apoyo individualizado según el campo visual y la agudeza. Se recomienda el uso de Smart Site (introducción a servicios locales de consulta de baja visión).
Consejo genético: Proporcionado por genetistas clínicos y consejeros genéticos certificados. Las consultas comunes incluyen estimación del riesgo de recurrencia, educación, empleo, matrimonio y parto. Se puede recibir consejo genético incluso sin someterse a pruebas genéticas.
Sistema de enfermedades intratables: Está disponible un subsidio de gastos médicos como enfermedad intratable designada 9). También considere obtener un certificado de discapacidad física y el apoyo médico para la independencia.
Voretigene neparvovec (Luxturna): Un fármaco de terapia génica que puede administrarse a pacientes con variantes patogénicas bialélicas en el gen RPE65 y suficientes células retinianas viables. Aprobado en Japón en 2023 9). En el ensayo de fase III de EE. UU. (301), se inscribieron 31 pacientes; el análisis mITT (20 intervención, 9 control) mostró una mejora significativa en MLMT y FST de luz blanca en comparación con el grupo control 18). En el ensayo de fase III nacional (A11301), se confirmó un aumento de la sensibilidad FST y una expansión del campo visual en 4 pacientes japoneses 19). La mejora de la agudeza visual es limitada, y se han reportado complicaciones a largo plazo como atrofia coriorretiniana en más del 20% de los pacientes 9).
Realice lo siguiente cada 6 meses a 1 año: agudeza visual, microscopía con lámpara de hendidura, fondo de ojo, campo visual Humphrey (HFA 10-2), OCT9).
Q¿Qué fármacos se utilizan para tratar el edema macular?
A
El tratamiento de primera línea para RP-CME son los inhibidores de la anhidrasa carbónica (IAC), utilizándose colirio de dorzolamida o acetazolamida oral 9). La mejora del CMT se logra en aproximadamente el 40% de los casos, pero se observa recurrencia en aproximadamente el 30%. Si el IAC no es efectivo, se consideran la inyección intravítrea de triamcinolona acetonida o el implante intravítreo de dexametasona (Ozurdex). Los fármacos anti-VEGF no se recomiendan para RP-CME. Cabe señalar que todos estos se usan fuera de indicación.
La vía común final de la muerte de los fotorreceptores en la RP es la apoptosis. Aunque los tipos de mutaciones genéticas son diversos, en última instancia convergen en una vía común de muerte celular.
En la RP, los bastones típicamente degeneran y desaparecen primero, seguidos de la degeneración secundaria de los conos 7). Los conos dependen de factores tróficos producidos por los bastones (RdCVF: Factor de Viabilidad de Conos Derivado de Bastones) para sobrevivir, por lo que tras la pérdida de bastones, los conos también pierden función 7)11).
La retina es uno de los tejidos con mayor actividad metabólica, convirtiendo el 80-90% de la glucosa en lactato mediante glucólisis aeróbica (efecto Warburg). Los conos son más vulnerables al estrés metabólico que los bastones, y esta vulnerabilidad metabólica también contribuye a la degeneración secundaria de conos 11).
La inflamación también se reconoce como un factor importante en la progresión de la RP, y la activación de microglía y la infiltración de macrófagos empeoran el daño retiniano 11). El estrés oxidativo también actúa como impulsor biológico de la degeneración secundaria de conos.
Los mecanismos de degeneración difieren según el gen.
Mutación RHO: La rodopsina mal plegada induce estrés del retículo endoplásmico → UPR (respuesta a proteínas desplegadas) → apoptosis11)
Mutaciones en REEP6: REEP6 codifica una proteína involucrada en el mantenimiento de la morfología del RE. Las mutaciones patogénicas provocan la formación de cuerpos de inclusión del RE en el segmento externo de los bastones, lo que lleva a la degeneración fotorreceptora 4)
Mutaciones en RPGR: RPGR participa en la estructura axonémica de los cilios primarios, y las mutaciones alteran el transporte de sustancias al segmento externo del fotorreceptor1)
7. Investigación más reciente y perspectivas futuras (informes en fase de investigación)
La terapia génica es el enfoque más prometedor para tratar enfermedades retinianas hereditarias 8).
Luxturna (voretigene neparvovec): Un fármaco de terapia génica para pacientes con variantes patogénicas bialélicas en el gen RPE65. En el ensayo de fase III en EE. UU. (estudio 301), se inscribieron 31 pacientes, y el análisis mITT (20 de intervención, 9 de control) mostró una mejora significativa en MLMT y FST de luz blanca en comparación con el grupo de control 18). En el ensayo de fase III nacional (estudio A11301), también se confirmó un aumento de la sensibilidad FST y una expansión del campo visual en 4 pacientes japoneses 19). Fue aprobado en Japón en 2023, sirviendo como puente entre el tratamiento estándar y la terapia en investigación.
Terapia génica para RPGR: La terapia génica mediada por AAV para la RP ligada al cromosoma X causada por mutaciones en RPGR ha avanzado a ensayos clínicos de fase I/II/III 8).
CRISPR/Cas9: Se están realizando investigaciones para la corrección directa de mutaciones patogénicas y la inactivación de mutaciones dominantes negativas 8).
RdCVF (Factor de viabilidad de conos derivado de bastones) y terapia de protección de conos
RdCVF es una proteína secretada por los bastones que mantiene la supervivencia de los conos 7)11). Se están llevando a cabo ensayos clínicos de terapia de protección de conos utilizando RdCVF, y se destaca como una estrategia de tratamiento independiente para preservar la función de los conos después de la degeneración de los bastones.
NAC es un fármaco que suprime el estrés oxidativo, y los ensayos de fase I han informado mejoría en la agudeza visual17). A partir de 2025, se están realizando ensayos de fase III.
Potencial reutilización de glucocorticoides (dexametasona)
Estudios recientes in vivo (modelo de ratón rd10) han demostrado que la dexametasona intravítrea protege los fotorreceptores de cono y el epitelio pigmentario de la retina11). Los glucocorticoides tienen un fuerte potencial de reutilización como terapia independiente de la mutación. Sin embargo, la evidencia actual se limita a modelos animales, y se necesita más validación para la aplicación clínica en humanos.
Trasplante de retina derivado de células iPS y retina artificial
Trasplante de retina derivado de células iPS: Se están realizando investigaciones sobre el trasplante de láminas de fotorreceptores generadas a partir de las propias células iPS del paciente.
Retina artificial (prótesis retiniana): Dispositivos de estimulación eléctrica para RP en etapa terminal. Argus II y otros se han comercializado en el extranjero, y en Japón están en curso ensayos clínicos del método de estimulación suprachoroidal transretiniana.
Q¿Está disponible la terapia génica en Japón?
A
Voretigene neparvovec (Luxturna) fue aprobado en Japón en 2023, pero solo para distrofia retiniana con variantes patogénicas bialélicas en el gen RPE65 9)18)19). La terapia génica para RP causada por otras mutaciones genéticas, incluidas las mutaciones RPGR, se encuentra actualmente en fase de ensayos clínicos 7) y no está aprobada como tratamiento general en Japón.
Q¿Cómo puedo recibir tratamientos en fase de investigación?
A
La participación en ensayos clínicos se limita a estudios formales aprobados por el comité de ética de la institución médica. Además de consultar con su médico, puede buscar información sobre ensayos en el Japan Registry of Clinical Trials (jRCT) operado por el Centro Nacional del Cáncer o en ClinicalTrials.gov de Estados Unidos.
Baz-Redón N, Sánchez-Bellver L, Fernández-Cancio M, Rovira-Amigo S, Burgoyne T, Ranjit R, Aquino V, Toro-Barrios N, et al. Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene. Cells. 2024;13(6). doi:10.3390/cells13060524. PMID:38534367; PMCID:PMC10968961.
Holanda IP, et al. Syndromic Retinitis Pigmentosa: A 15-Patient Study Exploring Clinical and Genetic Features. Genes. 2024;15(4):516.
Wawrocka A, Walczak-Sztulpa J, Kuszel L, Niedziela-Schwartz Z, Skorczyk-Werner A, Bernardczyk-Meller J, Krawczynski MR. Coexistence of Retinitis Pigmentosa and Ataxia in Patients with PHARC, PCARP, and Oliver-McFarlane Syndromes. International journal of molecular sciences. 2024;25(11). doi:10.3390/ijms25115759. PMID:38891946; PMCID:PMC11172263.
Lujia Zhang, Ya Li, Litao Qin, Yu Wu, Bo Lei. Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population. Genes. 2021;12(4):537. doi:10.3390/genes12040537.
Daiki Sakai, Seiji Takagi, Yasuhiko Hirami, Makoto Nakamura, Yasuo Kurimoto. Use of ellipsoid zone width for predicting visual prognosis after cataract surgery in patients with retinitis pigmentosa. Eye. 2022;37(1):42-47. doi:10.1038/s41433-021-01878-3.
日本眼科学会. 網膜色素変性診療ガイドライン. 日眼会誌. 2016;120:846-861.
Campochiaro PA, Mir TA. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res. 2018;62:24-37. doi:10.1016/j.preteyeres.2017.08.004.
Liu Y, Zong X, Cao W, Zhang W, Zhang N, Yang N. Gene therapy for retinitis pigmentosa: current challenges and new progress. Biomolecules. 2024;14(8):903. PMID: 39199291. PMCID: PMC11352491. doi:10.3390/biom14080903.
Napoli D, Di Marco B, Salamone G, Orsini N, Mazziotti R, Strettoi E. Keeping the lights on: a new role for an old drug to support cone survival in Retinitis Pigmentosa. Progress in retinal and eye research. 2025;109:101403. doi:10.1016/j.preteyeres.2025.101403. PMID:40998215.
Koyanagi Y, Akiyama M, Nishiguchi KM, Momozawa Y, Kamatani Y, Takata S, et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. Journal of medical genetics. 2019;56(10):662-670. doi:10.1136/jmedgenet-2018-105691. PMID:31213501.
Numa S, Oishi A, Higasa K, Oishi M, Miyata M, Hasegawa T, Ikeda HO, Otsuka Y, Matsuda F, Tsujikawa A.. EYS is a major gene involved in retinitis pigmentosa in Japan: genetic landscapes revealed by stepwise genetic screening. Sci Rep. 2020;10(1):20770. doi:10.1038/s41598-020-77558-1. PMID:33247286; PMCID:PMC7695703.
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, Willett W.. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. doi:10.1001/archopht.1993.01090060049022. PMID:8512476.
Nakazawa M, Suzuki Y, Ito T, Metoki T, Kudo T, Ohguro H. Long-term effects of nilvadipine against progression of the central visual field defect in retinitis pigmentosa: an extended study. BioMed research international. 2013;2013:585729. doi:10.1155/2013/585729. PMID:24319686; PMCID:PMC3844269.
Yamamoto S, Sugawara T, Murakami A, Nakazawa M, Nao-I N, Machida S, et al. Topical isopropyl unoprostone for retinitis pigmentosa: microperimetric results of the phase 2 clinical study. Ophthalmology and therapy. 2012;1(1):5. doi:10.1007/s40123-012-0005-9. PMID:25135585; PMCID:PMC4108136.
Campochiaro PA, Iftikhar M, Hafiz G, Akhlaq A, Tsai G, Wehling D, et al. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. The Journal of clinical investigation. 2020;130(3):1527-1541. doi:10.1172/JCI132990. PMID:31805012; PMCID:PMC7269599.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.