El acromatopsia (ACHM) es una enfermedad retiniana hereditaria bilateral poco frecuente en la que los tres tipos de fotorreceptores conos pierden su función. También se denomina monocromatismo de bastones o ceguera total al color 1).
La prevalencia mundial se estima en aproximadamente 1 de cada 30,000 personas 1). Sigue un patrón de herencia autosómico recesivo, no se asocia con anomalías sistémicas y la esperanza de vida es normal.
El ACHM tiene dos formas: completa e incompleta. En la forma completa, toda la función de los conos está ausente; en la forma incompleta, al menos un subtipo de cono conserva cierta función, con una agudeza visual de alrededor de 20/40 a 20/120 y fotofobia y nistagmo más leves 2).
Se han identificado seis genes causantes (CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, ATF6), y el gen causante puede identificarse en más del 90% de los casos 1)2). Solo CNGA3 y CNGB3 representan el 80-90% de todos los casos.
Tenga en cuenta que las deficiencias comunes de la visión del color (anomalías congénitas de la visión del color) son causadas por anomalías en uno o dos tipos de fotopigmentos de los conos y afectan solo a la visión del color. La ACHM es fundamentalmente diferente porque todos los conos no funcionan, acompañada de disminución de la agudeza visual, nistagmo y fotofobia.
Hay un caso conocido de efecto fundador en el atolón de Pingelap (Micronesia). Después de que un tifón en el siglo XVIII redujera drásticamente la población, la mutación CNGB3 (p.S435F) se extendió entre los isleños, alcanzando una prevalencia de aproximadamente el 10% y una tasa de portadores de aproximadamente el 30%1)2).
Q¿En qué se diferencia la acromatopsia de la deficiencia común de la visión del color (debilidad cromática)?
A
Las anomalías congénitas comunes de la visión del color son causadas por anomalías en uno o dos tipos de fotopigmentos de los conos, afectando solo la visión del color, con agudeza visual normal. La acromatopsia implica la pérdida de función de los tres tipos de conos, por lo que además de la pérdida de la visión del color, se diferencia fundamentalmente por estar acompañada de disminución de la agudeza visual (aproximadamente 0.1 o menos), nistagmo, fotofobia y hemeralopía.
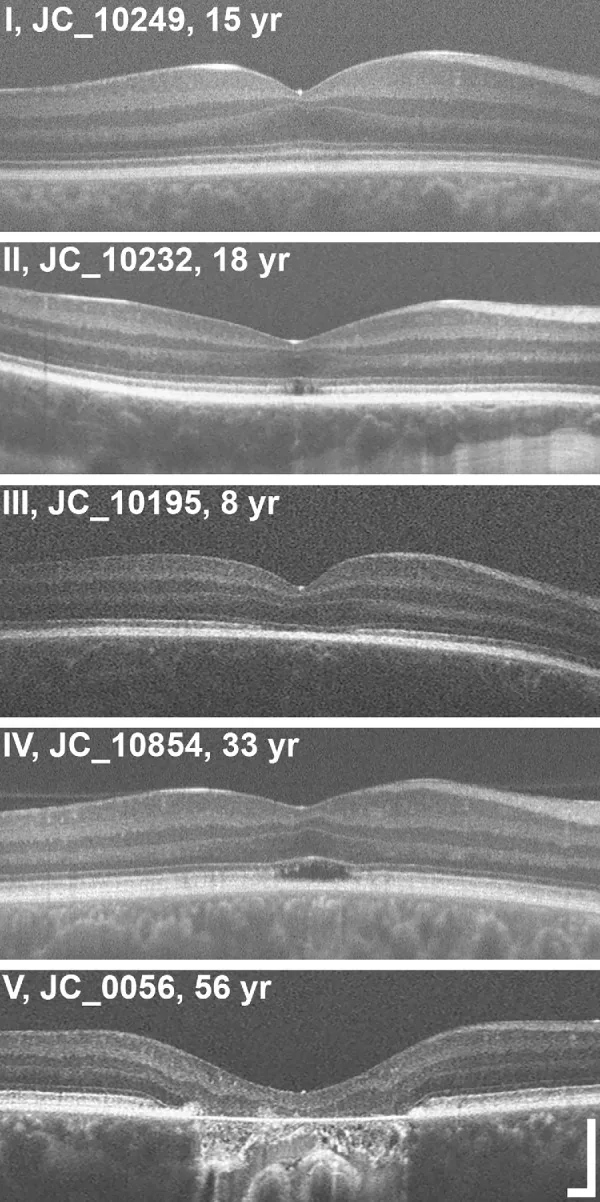
Grissim G, et al. Longitudinal Assessment of OCT-Based Measures of Foveal Cone Structure in Achromatopsia. Invest Ophthalmol Vis Sci. 2024. Figure 1. PMCID: PMC11005076. License: CC BY.
Clasificación de cinco grados de EZ en OCT, que muestra EZ normal (I), interrupción de EZ (II), pérdida de EZ (III), zona hiporreflectiva (IV) y atrofia retiniana externa con EPR (V). Esto corresponde a las anomalías de la zona elipsoide discutidas en la sección “2. Síntomas principales y hallazgos clínicos.”
Los síntomas de la ACHM comienzan a aparecer dentro de las primeras semanas después del nacimiento.
Disminución de la agudeza visual: En el tipo completo, 20/200 (aproximadamente 0.1) o menos; en el tipo incompleto, aproximadamente 20/80 (aproximadamente 0.25).
Fotofobia (sensibilidad a la luz): El 38% de los pacientes en una encuesta la reportaron como el síntoma más significativo2). La función visual disminuye notablemente en entornos brillantes.
Hemeralopía (ceguera diurna): La función visual disminuye con luz brillante, mientras que se mantiene una visión relativamente buena en entornos oscuros.
Deficiencia/pérdida de la visión cromática: Pérdida completa de la visión del color en los tres ejes1). Las láminas de Ishihara son casi ilegibles excepto la de demostración. La prueba Panel D-15 muestra líneas de confusión oblicuas circulares.
Nistagmo (nistagmo pendular): Aparece dentro de las primeras semanas de vida. Tiende a mejorar con el crecimiento y disminuye durante la visión cercana.
Hipermetropía asociada: La hipermetropía es común, pero también pueden presentarse un amplio rango de errores refractivos, incluida la miopía.
Reflejo pupilar paradójico: Hallazgo característico de constricción pupilar inicial en la oscuridad. Una pista importante para el diagnóstico en niños.
Hallazgos de fondo de ojo: Inicialmente parece casi normal. La angiografía fluoresceínica también muestra hallazgos casi normales. Con el tiempo, pueden ocurrir cambios parcheados y atrofia del epitelio pigmentario de la retina (EPR). A menudo se observan ausencia del reflejo foveal y degeneración macular. La OCT muestra una zona elipsoide y una zona de interdigitación irregulares e indistintas de la retina externa.
Hallazgos del electrorretinograma: El ERG fotópico muestra respuestas de conos marcadamente reducidas o ausentes, mientras que el ERG escotópico muestra respuestas de bastones normales o casi normales1)2). En el tipo GNAT2, los conos S están relativamente preservados en comparación con el tipo CNGA3/CNGB3.
Hipoplasia foveal: Presente en el 60–70% de los casos con mutaciones CNGA3/CNGB32).
FAF (Autofluorescencia del fondo de ojo): Existen cuatro patrones: normal, aumento de la señal central, disminución de la señal central, y anillo hiperfluorescente con hipofluorescencia central2).
Estadificación por OCT: Los cambios estructurales en las capas externas de la fóvea se evalúan en 5 etapas3).
Preservación de las capas retinianas externas (hiperreflectividad de la MLI, aplanamiento de la EZ)
Estadio 2
Disrupción de la zona elipsoide (EZ)
Etapa 3
Aparición de espacio ópticamente vacío
Etapa 4
Espacio ópticamente vacío + destrucción parcial del EPR
Etapa 5
Pérdida de la capa nuclear externa y/o destrucción completa del EPR
En un estudio de seguimiento de 10 años, a pesar de la agudeza visual mejor corregida estable (20/400 a 20/200), se confirmó la progresión estructural en la OCT (expansión del espacio ópticamente vacío: ojo derecho 246×59 μm, ojo izquierdo 326×53 μm)3).
Los focos hiperreflectivos aparecen más de 3 años antes de los cambios en la zona elipsoide, lo que sugiere que podrían ser marcadores tempranos de progresión de la enfermedad3).
AOSLO (Oftalmoscopia de barrido láser con óptica adaptativa): El mosaico de conos muestra espacios oscuros, aumento del espaciado de conos y disminución de la densidad de conos. No hay diferencias significativas entre los tipos CNGA3 y CNGB3; el tipo GNAT2 muestra conos relativamente preservados2).
Pruebas de visión cromática: Las láminas de Ishihara son casi ilegibles excepto la lámina de demostración. El Panel D-15 muestra un patrón de error de eje escotópico (entre deuteran y tritan). El anomaloscopio muestra una pendiente pronunciada y no incluye el rango de igualación normal.
Q¿Empeora la agudeza visual en la acromatopsia con la edad?
A
La agudeza visual mejor corregida suele ser estable a largo plazo. Sin embargo, la evaluación estructural mediante OCT puede mostrar cambios progresivos (disrupción de la zona elipsoide, expansión del espacio ópticamente vacío) con el tiempo3). Es característica una disociación entre función y estructura, y es importante un monitoreo regular.
Herencia autosómica recesiva. Si ambos padres son portadores, el riesgo de tener un hijo afectado es del 25%1). Los afectados suelen nacer de padres sanos y a menudo no hay antecedentes familiares.
También se han reportado patrones de herencia no mendeliana debido a disomía uniparental (UPD) paterna. En un caso donde la homocigosis para CNGA3 c.778G>C (p.D260H) resultó de UPD, no se detectó mutación en la madre 6). Tales ejemplos tienen implicaciones importantes para la evaluación del riesgo de recurrencia en el asesoramiento genético.
Frecuencia: Aproximadamente 25–50% de los casos 1)2)
Patrón de mutación: Principalmente mutaciones de cambio de sentido. El dominio transmembrana S4 es un punto caliente.
Distribución geográfica: CNGA3 representa más del 80% en Oriente Medio y China.
CNGB3
Cromosoma: 8q21.3
Función: Subunidad beta del canal CNG
Frecuencia: Aproximadamente 50% de los casos 1)2)
Patrón de mutación: Principalmente mutaciones sin sentido, de cambio de marco y de empalme. c.1148delC es la mutación más frecuente.
Distribución geográfica: CNGB3 representa más del 50% en Europa y Estados Unidos.
Otros genes
GNAT2 (1p13.3): Transducina alfa de cono. Aproximadamente 2%. Relativamente leve, con preservación de la capa fotorreceptora 2)
PDE6C (10q23.33): Subunidad alfa de PDE de cono. Forma grave de inicio temprano 5)
PDE6H (12p12.3): Subunidad gamma de PDE de cono. Extremadamente raro 1)
ATF6 (1q23.3): Factor de transcripción de respuesta al estrés del retículo endoplásmico. Aproximadamente 2%. Mecanismo no directamente involucrado en la fototransducción 1)2)
Alelo hipomórfico: CNGB3 c.1208G>A (p.R403Q) retiene función parcial, resultando en un fenotipo leve 2).
Características de las mutaciones en PDE6C: En cuatro casos con mutaciones nuevas (c.1670G>A, c.2192G>A), se observó la tríada de nistagmo, fotofobia y trastornos de la visión cromática en todos. El electrorretinograma mostró ausencia de respuestas fotópicas y de flicker a 30 Hz con respuestas escotópicas normales, y los heterocigotos compuestos presentaron un fenotipo más grave 5).
Herencia digénica: Existen casos raros con mutaciones tanto en CNGA3 como en CNGB3 1).
Q¿Por qué un hijo puede desarrollar acromatopsia aunque ambos padres no la tengan?
A
Debido a que el acromatopsia es autosómico recesivo, ambos padres pueden ser portadores (cada uno con una copia del gen mutado) y parecer sanos con visión cromática normal. Cuando dos portadores tienen un hijo, hay un 25% de probabilidad de que el hijo herede dos copias de la mutación y desarrolle la enfermedad 1).
Si se observan nistagmo, fotofobia y disminución de la agudeza visual dentro de las primeras semanas de vida, es necesaria una evaluación integral con enfoque en ACHM. La prueba genética es esencial para un diagnóstico definitivo.
Electrorretinograma
Importancia diagnóstica: Estándar de oro
Hallazgos del tipo completo: El electrorretinograma fotópico muestra respuestas de conos ausentes o gravemente reducidas. El electrorretinograma escotópico muestra respuestas de bastones normales o casi normales1)2)
Hallazgos del tipo incompleto: Se detectan respuestas de conos débiles correspondientes a la función de conos residual
Características del tipo GNAT2: Los conos S están relativamente preservados en comparación con el tipo CNGA3/CNGB3
OCT
Importancia diagnóstica: Estándar para la evaluación estructural
Contenido de la evaluación: Estadificación de 5 niveles de las capas externas de la fóvea, detección de hipoplasia foveal2)3)
Seguimiento: Incluso si es funcionalmente estable, puede ocurrir progresión estructural, por lo que el monitoreo regular es importante
Pruebas auxiliares: Evaluación multimodal combinando FAF y AOSLO
Prueba genética
Importancia diagnóstica: Esencial para el diagnóstico definitivo
Método: Panel dirigido de 6 genes (CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)2)
Tasa de resolución: Se puede identificar el gen causante en más del 90% de los casos1)
Participación en ensayos clínicos: El diagnóstico molecular es obligatorio para participar en ensayos clínicos de terapia génica
Es importante diferenciar la ACHM de enfermedades con presentaciones clínicas similares2).
Monocromatismo de conos azules (BCM): Herencia ligada al cromosoma X. Función del cono S parcialmente preservada. Se puede diferenciar con el anomaloscopio.
Distrofia de conos: Progresiva después del inicio. Se distingue por el patrón del electrorretinograma y la evolución.
Síndrome de Alström: Distrofia combinada de conos y bastones con síntomas sistémicos (obesidad, pérdida auditiva, etc.).
La hipermetropía es común, pero se asocia con una amplia gama de errores refractivos. La corrección con gafas o lentes de contacto es importante para maximizar la agudeza visual. Si hay ambliopía, se puede considerar la oclusión o el tratamiento con atropina.
Las lentes tintadas para reducir la fotofobia mejoran significativamente la calidad de vida. Encuestas a pacientes muestran que el 96% prefiere los filtros grises sobre los rojos, y el 74% prefiere los filtros grises al aire libre2). La selección debe adaptarse a las preferencias individuales y al entorno de actividad del paciente.
Se necesita explicación y apoyo de expertos sobre las características de la herencia autosómica recesiva, el riesgo de recurrencia y la importancia del diagnóstico genético. También se recomienda someterse a un diagnóstico genético para prepararse para futuros ensayos clínicos de terapia génica.
Q¿Qué color de gafas de filtro de luz es adecuado?
A
En una encuesta de pacientes, el 96% prefirió los filtros grises sobre los rojos, y el 74% prefirió los filtros grises en exteriores2). Sin embargo, debido a las diferencias individuales, es recomendable probar varios filtros y elegir el que se adapte a su entorno de actividad en consulta con un oftalmólogo o especialista en baja visión.
6. Fisiopatología y mecanismo detallado de la enfermedad
La cascada de fototransducción en los conos normales es la siguiente1).
Estado oscuro: La concentración intracelular de cGMP es alta, los canales CNG están abiertos, la entrada de Na⁺ y Ca²⁺ mantiene la despolarización y se libera glutamato de forma continua.
Exposición a la luz: La opsina (pigmento visual) se activa → la transducina (proteína G) se activa → la fosfodiesterasa (PDE) se activa → el cGMP se hidroliza → los canales CNG se cierran → hiperpolarización → se suprime la liberación de glutamato.
Retroalimentación negativa: GCAP (proteína activadora de guanilato ciclasa) se une al Ca²⁺ e inhibe la actividad de retGC, regulando la producción de cGMP.
Mutaciones en CNGA3: Las mutaciones sin sentido alteran el plegamiento, el tráfico intracelular y la integración en la membrana de la proteína1)7). El dominio transmembrana S4 es un punto caliente de mutaciones. Se han notificado más de 150 mutaciones sin sentido; 103 eran de patogenicidad incierta, pero el análisis estructura-función 3D sugiere que el 86.4% tiene consecuencias funcionales similares a mutaciones patogénicas conocidas7).
Mutaciones en CNGB3: Las mutaciones sin sentido y de cambio de marco producen proteínas de canal truncadas o sin función1). En la deficiencia de CNGB3, los canales homoméricos residuales de CNGA3 pueden preservar algo de función de los conos.
Mutaciones en ATF6: ATF6 es un factor de transcripción involucrado en la respuesta a proteínas mal plegadas (UPR) del retículo endoplásmico y no participa directamente en la cascada de fototransducción1)2). Su mecanismo patogénico difiere del de otros genes y aún está bajo investigación.
El canal CNG tiene una estructura tetramérica (CNGA3 × 3 y CNGB3 × 1, o 2:2 en algunos informes), y cada subunidad contiene seis dominios transmembrana, un dominio de unión a nucleótidos cíclicos, una región C-linker y un dominio formador del poro1).
Mecanismo y progresión de la degeneración de conos
El desarrollo y la morfología postnatal de los conos son casi normales, y se cree que la degeneración comienza en la edad adulta temprana. La acumulación de cGMP está involucrada en el proceso degenerativo, y los modelos animales muestran una progresión más rápida en las regiones ricas en conos S1).
Tradicionalmente, la ACHM se consideraba una enfermedad no progresiva. Sin embargo, la observación a largo plazo con OCT ha demostrado que los cambios estructurales (disrupción de la EZ, agrandamiento de espacios ópticos) progresan a pesar de que la mejor agudeza visual corregida permanece casi estable3). Esta disociación funcional-estructural tiene implicaciones importantes para evaluar la ventana terapéutica de la terapia génica.
En la zona libre de bastones de la fóvea (área densa en conos sin bastones), la pérdida de conos es prominente, mientras que en la parafóvea, los bastones contribuyen a la banda EZ, compensando la función 3).
7. Investigación más reciente y perspectivas futuras (informes en fase de investigación)
En el ensayo NCT03001310 (AAV8-hCARp.hCNGB3) con 11 adultos y 12 niños (23 participantes en total), la seguridad fue aceptable. Se observó mejoría en la visión del color en 6/23, mejoría en la fotofobia en 11/20 y mejoría en la calidad de vida (QoL) en 21/23. También se observó una tendencia al aumento de la inflamación intraocular con dosis altas 2).
En el ensayo de AAV8.CNGA3 (RD-CURE), 9 participantes recibieron tres dosis (1×10¹⁰ a 1×10¹¹ vg/ojo). A los 1 y 3 años, se observaron tendencias de mejora en la agudeza visual y la sensibilidad al contraste, pero no alcanzaron significación estadística. La seguridad fue buena 1)2).
McKyton et al. (2021) realizaron un mapeo visual cortical mediante fMRI después de la inyección subretiniana de AAV2tYF-PR1.7-hCNGA3 (NCT02935517) en dos adultos con CNGA3-ACHM 4). El ojo tratado mostró una tolerancia a la luz 5 veces mayor (mejoría dramática de la fotofobia) en comparación con el ojo no tratado, adquisición de la capacidad de detección del rojo y reducción del tamaño del campo receptivo poblacional (pRF) (lo que sugiere una mejora en la resolución espacial). Sin embargo, no se observó activación de áreas corticales específicas del color (p. ej., V4), y las respuestas de los conos permanecieron indetectables en el electrorretinograma de campo completo. Los pacientes informaron mejoras en la vida diaria, como “mayor seguridad al cruzar la calle”, “no necesitar lupas” y “no necesitar gafas de sol al aire libre”.
Se ha confirmado la recuperación funcional después de la suplementación génica en múltiples modelos animales 1)2).
Modelo de ratón: Recuperación hasta el 80% de lo normal en electrorretinograma.
Modelo canino (CNGB3): Recuperación del electrorretinograma de parpadeo de conos a los 2.5 años de seguimiento después de la suplementación génica. Mejora del comportamiento en entornos con más de 25 lux.
Modelo ovino (CNGA3): Mejora a largo plazo durante al menos 6 años después de la suplementación génica.
Primates no humanos (PDE6C): Recuperación funcional confirmada.
En cuanto a la ventana de tiempo terapéutica, los estudios en animales han demostrado que el tratamiento a una edad más temprana es más efectivo. Los ratones viejos mostraron una respuesta deficiente, pero el pretratamiento con CNTF (factor neurotrófico ciliar) permitió la recuperación funcional incluso en perros viejos 1)2).
Está en curso un ensayo clínico (NCT04041232) de fenilbutirato de glicerol (PBA) dirigido a la respuesta al estrés del retículo endoplásmico 2). Este enfoque está atrayendo la atención como un método para aliviar el plegamiento anómalo en el retículo endoplásmico en lugar de la suplementación génica.
Los principales desafíos de la terapia génica son los siguientes 1)2)4).
Inmunogenicidad: La respuesta inmune al cápside de AAV puede limitar la eficacia terapéutica.
Optimización de la dosis: Es necesario equilibrar el efecto terapéutico y el riesgo de inflamación intraocular.
Desafíos del desarrollo: Dificultad de la cirugía en ojos pediátricos y manejo de la ambliopía.
Limitaciones de la plasticidad de la corteza visual: La reorganización funcional cortical puede ser difícil en adultos, por lo que el momento del tratamiento es importante.
Eficacia a largo plazo y costo: Garantizar un efecto sostenido y desafíos de economía de la salud.
Q¿Puede la terapia génica curar completamente el acromatopsia?
A
Actualmente se encuentra en la fase de ensayos de seguridad y eficacia de fase I/II. Se ha informado reducción de la fotofobia y cierta mejora en la sensibilidad a la luz, pero no se ha logrado una recuperación completa de la visión cromática2)4). Estudios en animales sugieren que el tratamiento a una edad temprana puede ser efectivo, pero los datos a largo plazo en humanos aún son limitados. Es importante monitorear el progreso de la investigación y actualizar la información regularmente con el médico tratante.
McKyton A, Averbukh E, Marks Ohana D, Levin N, Banin E. Cortical Visual Mapping following Ocular Gene Augmentation Therapy for Achromatopsia. J Neurosci. 2021;41(35):7363-7371. doi:10.1523/JNEUROSCI.3222-20.2021. PMID:34349002; PMCID:PMC8412991.
Madeira C, Godinho G, Grangeia A, Falcão M, Silva R, Carneiro Â, et al. Two Novel Disease-Causing Variants in the PDE6C Gene Underlying Achromatopsia. Case reports in ophthalmology. 2021;12(3):749-760. doi:10.1159/000512284. PMID:34720973; PMCID:PMC8460892.
Kohl S, Baumann B, Dassie F, et al. Paternal Uniparental Isodisomy of Chromosome 2 in a Patient with CNGA3-Associated Autosomal Recessive Achromatopsia. Int J Mol Sci. 2021;22(15):7842. doi:10.3390/ijms22157842. PMID:34360608; PMCID:PMC8346044.
Rasmussen DK, Sun YJ, Franco JA, et al. Structure-function analysis of CNGA3-associated achromatopsia patient variants complements clinical genomics in pathogenicity determination. Orphanet J Rare Dis. 2025;20:261. doi:10.1186/s13023-025-03792-3.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.