A acromatopsia (ACHM) é uma doença hereditária rara da retina, bilateral, na qual os três tipos de cones perdem sua função. Também é chamada de “monocromatismo de bastonetes” ou “daltonismo total” 1).
A prevalência é estimada em cerca de 1 em 30.000 pessoas no mundo 1). A herança é autossômica recessiva, sem anormalidades sistêmicas associadas e expectativa de vida normal.
Existem dois tipos de ACHM: completa e incompleta. Na completa, há ausência total da função dos cones; na incompleta, pelo menos um subtipo de cone mantém função residual, com acuidade visual em torno de 20/40 a 20/120, e fotofobia e nistagmo mais leves 2).
Seis genes causadores são conhecidos (CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, ATF6), e o gene causador pode ser identificado em mais de 90% dos casos 1)2). Apenas CNGA3 e CNGB3 respondem por 80-90% dos casos.
Note-se que as anomalias gerais da visão de cores (discromatopsias congênitas) são causadas por anormalidades em um ou dois pigmentos visuais dos cones e afetam apenas a visão de cores. A ACHM difere fundamentalmente porque todos os cones não funcionam, acompanhada de baixa acuidade visual, nistagmo e fotofobia.
Há um caso conhecido como efeito fundador na Ilha de Pingelap (Micronésia). Após um tufão no século XVIII, a população da ilha diminuiu drasticamente, e a mutação CNGB3 (p.S435F) se espalhou entre os sobreviventes, atingindo uma prevalência de cerca de 10% e uma taxa de portadores de cerca de 30%1)2).
QQual a diferença entre acromatopsia e discromatopsia geral (daltonismo)?
A
As discromatopsias congênitas gerais são causadas por anormalidades em um ou dois pigmentos visuais dos cones e afetam apenas a visão de cores, com acuidade visual normal. Na acromatopsia, a função de todos os três cones está ausente, resultando em perda da visão de cores, além de baixa acuidade visual (cerca de 0,1 ou menos), nistagmo, fotofobia e hemeralopia, diferindo fundamentalmente.
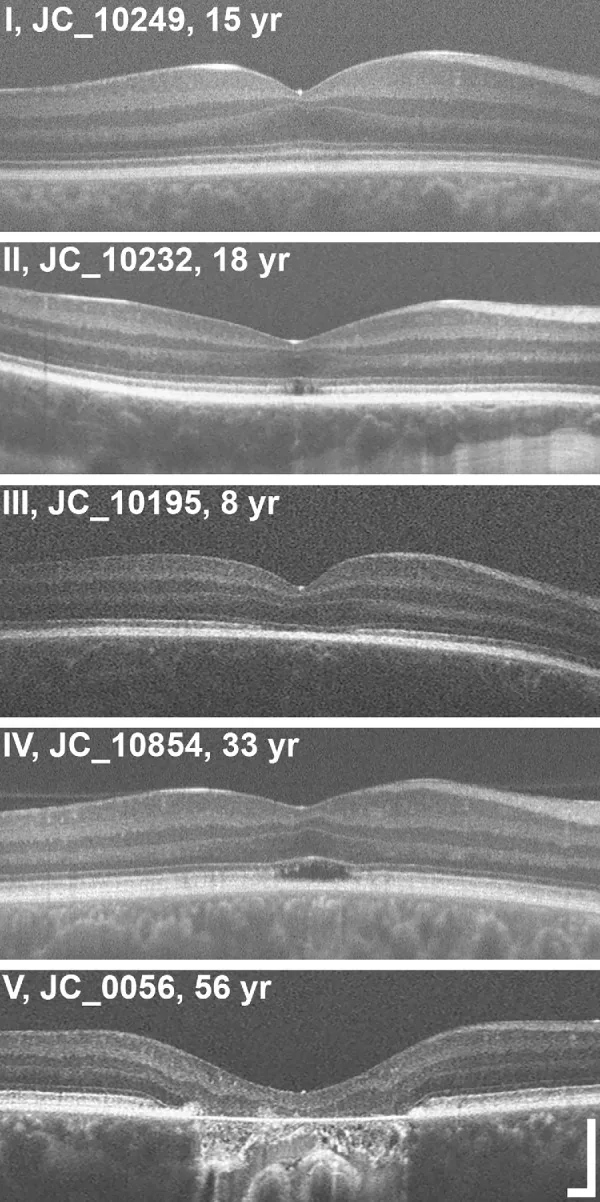
Grissim G, et al. Longitudinal Assessment of OCT-Based Measures of Foveal Cone Structure in Achromatopsia. Invest Ophthalmol Vis Sci. 2024. Figure 1. PMCID: PMC11005076. License: CC BY.
Classificação de cinco graus de EZ por OCT, mostrando EZ normal (I), descontinuidade da EZ (II), desaparecimento da EZ (III), área de baixa refletividade (IV) e atrofia da retina externa e EPR (V). Corresponde às anormalidades da zona elipsoide discutidas na seção “2. Principais sintomas e achados clínicos”.
Os sintomas da ACHM começam a aparecer nas primeiras semanas após o nascimento.
Baixa acuidade visual: No tipo completo, abaixo de 20/200 (cerca de 0,1); no tipo incompleto, cerca de 20/80 (cerca de 0,25).
Fotofobia (sensibilidade à luz): Em uma pesquisa com pacientes, 38% relataram como o sintoma mais significativo2). A função visual diminui acentuadamente em ambientes claros.
Hemeralopia (cegueira diurna): A função visual diminui em ambientes claros, mas é relativamente boa em ambientes escuros.
Perda/deficiência de visão de cores: Perda de visão de cores em todos os três eixos1). No teste de Ishihara, quase todas as placas são ilegíveis, exceto a placa de demonstração. O teste de painel D-15 mostra linhas de confusão circulares oblíquas.
Nistagmo pendular: Aparece nas primeiras semanas após o nascimento. Tende a melhorar com o crescimento e diminui na visão de perto.
Resposta pupilar paradoxal: Constrição pupilar inicial no escuro. Achado característico importante no diagnóstico infantil.
Achados de fundo de olho: Inicialmente quase normais. A angiografia fluoresceínica também mostra achados quase normais. Com o tempo, podem ocorrer alterações em manchas e atrofia do epitélio pigmentar da retina. Frequentemente observa-se ausência do reflexo foveal ou degeneração macular. Na OCT, a junção entre os segmentos interno e externo dos fotorreceptores e a zona de interdigitação são irregulares e pouco nítidas.
Achados de eletrorretinograma: No ERG fotópico, a resposta dos cones está acentuadamente reduzida ou ausente, enquanto a resposta dos bastonetes no ERG escotópico é normal ou quase normal1)2). No tipo GNAT2, os cones S são relativamente mais preservados do que nos tipos CNGA3/CNGB3.
Hipoplasia foveal: Presente em 60-70% dos casos com mutações CNGA3/CNGB32).
FAF (Autofluorescência de Fundo): Existem quatro padrões: normal, aumento do sinal central, diminuição do sinal central, anel hiperfluorescente com hipofluorescência central2).
Estadiamento por OCT: Avaliação das alterações estruturais das camadas externas da fóvea em cinco estágios3).
Estágio
Achado de OCT
Estágio 1
Preservação das camadas externas da retina (hiperrefletividade da ELM, achatamento da EZ)
Estágio 2
Destruição da zona elipsoide (EZ)
Estágio 3
Aparecimento de espaço óptico vazio (optically empty space)
Estágio 4
Espaço óptico vazio + destruição parcial do EPR
Estágio 5
Desaparecimento da camada nuclear externa e/ou destruição completa do EPR
Em um estudo de acompanhamento de 10 anos, apesar da acuidade visual melhor corrigida estável (20/400 a 20/200), a progressão estrutural na OCT (expansão do espaço óptico vazio: olho direito 246×59 μm, olho esquerdo 326×53 μm) foi confirmada3).
Focos hiperrefletivos (hyperreflective foci) podem aparecer mais de 3 anos antes das alterações na EZ, sugerindo potencial como marcadores precoces de progressão da doença3).
AOSLO (Oftalmoscópio de Varredura a Laser com Óptica Adaptativa): O mosaico de cones mostra espaços escuros (dark space), aumento do espaçamento entre cones e diminuição da densidade de cones. Não há diferença significativa entre os tipos CNGA3 e CNGB3, enquanto o tipo GNAT2 preserva relativamente os cones2).
Teste de visão de cores: Nas tabelas de Ishihara, quase todas as tabelas são ilegíveis, exceto a tabela de demonstração. No teste Panel D-15, mostra um padrão de erros no eixo escotópico (entre deutan e tritan). No anomaloscópio, mostra uma inclinação acentuada e não inclui a faixa de igualdade de cores normal.
QA acuidade visual no daltonismo total piora com a idade?
A
A acuidade visual melhor corrigida geralmente permanece estável a longo prazo. Por outro lado, a avaliação estrutural por OCT pode mostrar alterações ao longo do tempo (destruição da zona elipsoide, expansão do espaço óptico vazio) que podem progredir3). Há uma dissociação entre função e estrutura, e o monitoramento regular por meio de exames periódicos é importante.
Herança autossômica recessiva. Se ambos os pais são portadores (carreadores), o risco de a criança desenvolver a doença é de 25%1). Frequentemente ocorre em filhos de pais aparentemente saudáveis e, em muitos casos, não há histórico familiar.
Um padrão de herança não mendeliano devido a dissomia uniparental (UPD) paterna também foi relatado. Em um caso em que um homozigoto para CNGA3 c.778G>C (p.D260H) foi estabelecido por UPD, nenhuma mutação foi detectada na mãe 6). Tais exemplos têm importância significativa na avaliação do risco de recorrência no aconselhamento genético.
Padrão de mutação: Mutações missense são predominantes. O domínio transmembrana S4 é um ponto crítico
Distribuição geográfica: No Oriente Médio e China, CNGA3 representa mais de 80%
CNGB3
Cromossomo: 8q21.3
Função: Subunidade beta do canal CNG
Frequência: Cerca de 50% dos casos1)2)
Padrão de mutação: Mutações nonsense, frameshift e de splicing são predominantes. c.1148delC é a mutação mais frequente
Distribuição geográfica: Na Europa e América, CNGB3 representa mais de 50%
Outros Genes
GNAT2 (1p13.3): Transducina α dos cones. Cerca de 2%. Relativamente leve com preservação da camada de fotorreceptores 2)
PDE6C (10q23.33): Subunidade α da PDE dos cones. Tipo grave de início precoce 5)
PDE6H (12p12.3): Subunidade γ da PDE dos cones. Muito raro 1)
ATF6 (1q23.3): Fator de transcrição de resposta ao estresse do retículo endoplasmático. Cerca de 2%. Mecanismo não diretamente envolvido na transdução de luz 1)2)
Alelo de baixa penetrância: CNGB3 c.1208G>A (p.R403Q) retém função parcial, resultando em fenótipo leve 2).
Características da mutação PDE6C: Em 4 casos com mutações novas (c.1670G>A, c.2192G>A), a tríade (nistagmo, fotofobia, distúrbio de visão de cores) foi observada em todos os casos. O eletrorretinograma mostrou ausência de visão fotópica e flicker de 30 Hz com visão escotópica normal, e heterozigotos compostos apresentaram fenótipo mais grave 5).
Herança digênica: Existem casos raros com mutações em ambos CNGA3 e CNGB3 1).
QPor que uma criança pode ter daltonismo total mesmo que os pais não tenham?
A
Como o daltonismo total é uma doença autossômica recessiva, ambos os pais podem ser portadores de uma cópia do gene mutante (portadores) sem sintomas e com visão de cores normal. Quando dois portadores têm filhos, a probabilidade de a criança herdar duas cópias da mutação e desenvolver a doença é de 25% 1).
Se nistagmo, fotofobia e baixa visão forem observados nas primeiras semanas após o nascimento, é necessário um exame abrangente considerando a possibilidade de ACHM. O teste genético é essencial para o diagnóstico definitivo.
Eletrorretinograma
Significado diagnóstico: Padrão ouro
Achados do tipo completo: Resposta dos cones no eletrorretinograma fotópico ausente ou drasticamente reduzida. Resposta dos bastonetes no eletrorretinograma escotópico normal a quase normal1)2)
Achados do tipo incompleto: Uma resposta cone fraca correspondente à função cone residual é detectada
Características do tipo GNAT2: Cones S relativamente mais preservados em comparação com o tipo CNGA3/CNGB3
OCT
Significado diagnóstico: Padrão para avaliação estrutural
Conteúdo da avaliação: Estadiamento de 5 estágios da camada externa da fóvea, detecção de hipoplasia foveal2)3)
Acompanhamento: Mesmo com estabilidade funcional, pode ocorrer progressão estrutural, portanto o monitoramento regular é importante
Exames auxiliares: Avaliação multimodal combinando FAF e AOSLO
Teste Genético
Significado diagnóstico: Essencial para o diagnóstico definitivo
Método: Painel direcionado para 6 genes (CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)2)
Taxa de resolução: O gene causador pode ser identificado em mais de 90% dos casos1)
Participação em ensaios clínicos: O diagnóstico molecular é obrigatório para participação em ensaios clínicos de terapia gênica
Embora a hipermetropia seja comum, há uma ampla gama de erros refrativos, portanto a correção com óculos ou lentes de contato é importante para maximizar a acuidade visual. Se houver ambliopia, considere terapia de oclusão ou atropina.
Lentes de proteção para reduzir a fotofobia melhoram significativamente a qualidade de vida diária. Dados de pesquisa com pacientes mostram que 96% preferem filtros cinza em vez de vermelhos, e 74% preferem filtros cinza ao ar livre2). A escolha adaptada às preferências individuais e ao ambiente de atividade do paciente é importante.
É necessária explicação e apoio de especialistas sobre as características da herança autossômica recessiva, risco de recorrência e importância do diagnóstico genético. Recomenda-se também realizar o diagnóstico genético para se preparar para futuros ensaios clínicos de terapia gênica.
QQuais cores são adequadas para óculos de proteção contra luz?
A
Em uma pesquisa com pacientes, 96% preferiram filtros cinza a filtros vermelhos, e 74% preferiram filtros cinza ao ar livre 2). No entanto, devido a diferenças individuais, é recomendável experimentar vários filtros e escolher o mais adequado ao seu ambiente de atividade em consulta com um oftalmologista ou especialista em baixa visão.
6. Fisiopatologia e Mecanismos Detalhados da Doença
A cascata de transdução de luz em células cone normais é a seguinte1).
No escuro: A concentração intracelular de cGMP é alta, os canais CNG estão abertos, Na⁺ e Ca²⁺ entram, a célula mantém um estado despolarizado e libera glutamato continuamente.
Na exposição à luz: A opsina (pigmento visual) é ativada → A transducina (proteína G) é ativada → A fosfodiesterase (PDE) é ativada → O cGMP é degradado → Os canais CNG fecham → Hiperpolarização → Inibição da liberação de glutamato.
Feedback negativo: A GCAP (Proteína Ativadora da Guanilato Ciclase) liga-se ao Ca²⁺ e inibe a atividade da retGC, regulando a produção de cGMP.
Mutação CNGA3: Mutações de sentido trocado prejudicam o dobramento da proteína, o transporte intracelular e a integração na membrana1)7). O domínio transmembrana S4 é um ponto crítico de mutação. Mais de 150 mutações de sentido trocado foram relatadas, 103 mutações tiveram patogenicidade não determinada, mas a análise estrutura-função 3D sugeriu que 86,4% têm consequências funcionais semelhantes a mutações patogênicas conhecidas7).
Mutação CNGB3: Mutações sem sentido e de deslocamento de quadro produzem proteínas de canal truncadas ou com perda de função1). Na deficiência de CNGB3, canais homoméricos CNGA3 permanecem, podendo preservar alguma função cone residual.
Mutação ATF6: É um fator de transcrição envolvido na resposta a proteínas mal dobradas (UPR) do retículo endoplasmático, e não participa diretamente da cascata de transdução de luz1)2). O mecanismo patológico difere de outros genes e ainda está sendo elucidado.
O canal CNG possui estrutura tetramérica (CNGA3 × 3 e CNGB3 × 1, em alguns relatos 2:2), cada subunidade tem 6 domínios transmembrana, um domínio de ligação a nucleotídeo cíclico, uma região conectora C e um domínio formador de poro1).
O desenvolvimento pós-natal principal dos cones e sua morfologia são quase normais, e a degeneração acredita-se que comece no início da idade adulta. O acúmulo de cGMP está envolvido no processo degenerativo, e uma progressão mais rápida em áreas ricas em cones S foi demonstrada em modelos animais1).
Tradicionalmente, a ACHM era considerada uma doença não progressiva. No entanto, a observação de longo prazo com OCT mostrou que, embora a melhor acuidade visual corrigida permaneça quase estável, alterações estruturais (destruição da EZ, alargamento de lacunas ópticas) progridem3). Essa dissociação função-estrutura é importante para avaliar a janela terapêutica da terapia gênica.
Na zona livre de bastonetes (área densa de cones) da fóvea, a perda de cones é proeminente, enquanto na parafóvea, os bastonetes compensam a função contribuindo para a banda EZ3).
7. Pesquisas Recentes e Perspectivas Futuras (Relatórios em Fase de Pesquisa)
No ensaio NCT03001310 (AAV8-hCARp.hCNGB3) com 11 adultos e 12 crianças (total de 23 indivíduos), a segurança foi aceitável. Melhora na visão de cores foi observada em 6/23, melhora na fotofobia em 11/20 e melhora na qualidade de vida em 21/23. Em doses altas, observou-se uma tendência de aumento da inflamação intraocular 2).
No ensaio AAV8.CNGA3 (RD-CURE), 9 indivíduos receberam três doses (1×10¹⁰ a 1×10¹¹ vg/olho). Dados de 1 e 3 anos mostraram tendência de melhora na acuidade visual e sensibilidade ao contraste, mas sem significância estatística. A segurança foi boa 1)2).
McKyton et al. (2021) realizaram mapeamento cortical por fMRI após injeção sub-retiniana de AAV2tYF-PR1.7-hCNGA3 (NCT02935517) em dois adultos com CNGA3-ACHM 4). O olho tratado mostrou tolerância à luz 5 vezes maior em comparação ao olho não tratado (melhora dramática da fotofobia), aquisição de capacidade de detecção de vermelho e redução do tamanho do campo receptivo populacional (pRF), sugerindo melhora na resolução espacial. Por outro lado, não foi observada ativação de áreas corticais específicas para cor (como V4), e o eletrorretinograma de campo total permaneceu incapaz de detectar respostas de cones. Os pacientes relataram melhorias na vida diária, como “maior sensação de segurança ao atravessar a rua”, “não precisar de lupa” e “não precisar de óculos escuros ao ar livre”.
A recuperação funcional após reposição gênica foi confirmada em vários modelos animais 1)2).
Modelo de camundongo: Recuperação de até 80% do normal no eletrorretinograma.
Modelo canino (CNGB3): Recuperação do eletrorretinograma de flicker de cone após 2,5 anos de acompanhamento pós-reposição gênica. Melhora comportamental em ambientes >25 lux.
Modelo ovino (CNGA3): Melhora de longo prazo por pelo menos 6 anos após reposição gênica.
Primatas não humanos (PDE6C): Confirmação de recuperação funcional.
Quanto à janela de tempo eficaz do tratamento, experimentos em animais mostraram que o tratamento em idade jovem é mais eficaz. Camundongos idosos apresentaram resposta ruim, mas o pré-tratamento com CNTF (fator neurotrófico ciliar) possibilitou a recuperação funcional em cães idosos 1)2).
Um ensaio clínico (NCT04041232) com fenilbutirato de glicerol (PBA) visando a resposta ao estresse do retículo endoplasmático está em andamento 2). Esta abordagem é notável como uma alternativa à reposição gênica, aliviando o dobramento incorreto no retículo endoplasmático.
Os principais desafios da terapia gênica são os seguintes 1)2)4).
Imunogenicidade: A resposta imune ao capsídeo do AAV pode limitar a eficácia do tratamento.
Otimização da dose: É necessário equilibrar a eficácia terapêutica e o risco de inflamação intraocular.
Desafios do desenvolvimento: Dificuldade da cirurgia em olhos de crianças, manejo do aparecimento de ambliopia.
Limites da plasticidade do córtex visual: Em adultos, a reorganização funcional do córtex pode ser difícil, tornando o momento do tratamento importante.
Eficácia a longo prazo e custo: Garantir efeito duradouro e desafios econômicos em saúde.
QA terapia gênica pode curar completamente o daltonismo total?
A
Atualmente, estamos na fase de ensaios de segurança e eficácia de fase I/II. A redução da fotofobia e alguma melhora na sensibilidade à luz foram relatadas, mas a recuperação total da visão de cores ainda não foi alcançada2)4). Estudos em animais sugerem que o tratamento em idade precoce pode ser eficaz, mas os dados de longo prazo em humanos ainda são limitados. É importante acompanhar o progresso da pesquisa e atualizar as informações regularmente com o médico responsável.
McKyton A, Averbukh E, Marks Ohana D, Levin N, Banin E. Cortical Visual Mapping following Ocular Gene Augmentation Therapy for Achromatopsia. J Neurosci. 2021;41(35):7363-7371. doi:10.1523/JNEUROSCI.3222-20.2021. PMID:34349002; PMCID:PMC8412991.
Madeira C, Godinho G, Grangeia A, Falcão M, Silva R, Carneiro Â, et al. Two Novel Disease-Causing Variants in the PDE6C Gene Underlying Achromatopsia. Case reports in ophthalmology. 2021;12(3):749-760. doi:10.1159/000512284. PMID:34720973; PMCID:PMC8460892.
Kohl S, Baumann B, Dassie F, et al. Paternal Uniparental Isodisomy of Chromosome 2 in a Patient with CNGA3-Associated Autosomal Recessive Achromatopsia. Int J Mol Sci. 2021;22(15):7842. doi:10.3390/ijms22157842. PMID:34360608; PMCID:PMC8346044.
Rasmussen DK, Sun YJ, Franco JA, et al. Structure-function analysis of CNGA3-associated achromatopsia patient variants complements clinical genomics in pathogenicity determination. Orphanet J Rare Dis. 2025;20:261. doi:10.1186/s13023-025-03792-3.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.