Die Achromatopsie (ACHM) ist eine seltene, beidseitige erbliche Netzhauterkrankung, bei der die Funktion aller drei Zapfentypen verloren geht. Sie wird auch als „Stäbchenmonochromasie“ oder „totale Farbenblindheit“ bezeichnet1).
Die Prävalenz wird weltweit auf etwa 1 von 30.000 geschätzt1). Die Vererbung erfolgt autosomal-rezessiv, es liegen keine systemischen Begleiterkrankungen vor, und die Lebenserwartung ist normal.
Es gibt zwei Formen der ACHM: komplette und inkomplette. Bei der kompletten Form fehlt die gesamte Zapfenfunktion; bei der inkompletten Form hat mindestens ein Zapfensubtyp eine Restfunktion, der Visus liegt bei etwa 20/40 bis 20/120, und Photophobie sowie Nystagmus sind milder ausgeprägt2).
Sechs Gene (CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, ATF6) sind ursächlich, und bei über 90 % der Fälle kann das verantwortliche Gen identifiziert werden1)2). Allein die beiden Gene CNGA3 und CNGB3 machen 80–90 % aller Fälle aus.
Beachten Sie, dass allgemeine Farbsehstörungen (angeborene Farbsehstörungen) auf eine Anomalie von ein oder zwei Arten von Zapfen-Sehpigmenten zurückzuführen sind und nur das Farbsehen betreffen. ACHM unterscheidet sich grundlegend, da alle Zapfen nicht funktionieren, was zu verminderter Sehschärfe, Nystagmus und Photophobie führt.
Es gibt einen bekannten Fall des Gründereffekts auf der Insel Pingelap (Mikronesien). Nach einem Taifun im 18. Jahrhundert verbreitete sich die CNGB3-Mutation (p.S435F) unter der stark dezimierten Bevölkerung, mit einer Prävalenz von etwa 10 % und einer Trägerrate von etwa 30 %1)2).
QWas ist der Unterschied zwischen Achromatopsie und allgemeinen Farbsehstörungen (Farbsehschwäche)?
A
Allgemeine angeborene Farbsehstörungen sind auf eine Anomalie von ein oder zwei Arten von Zapfen-Sehpigmenten zurückzuführen, betreffen nur das Farbsehen, und die Sehschärfe ist normal. Bei der Achromatopsie fehlt die Funktion aller drei Zapfentypen, was zusätzlich zum Verlust des Farbsehens zu verminderter Sehschärfe (etwa 0,1 oder weniger), Nystagmus, Photophobie und Hemeralopie führt, was sie grundlegend unterscheidet.
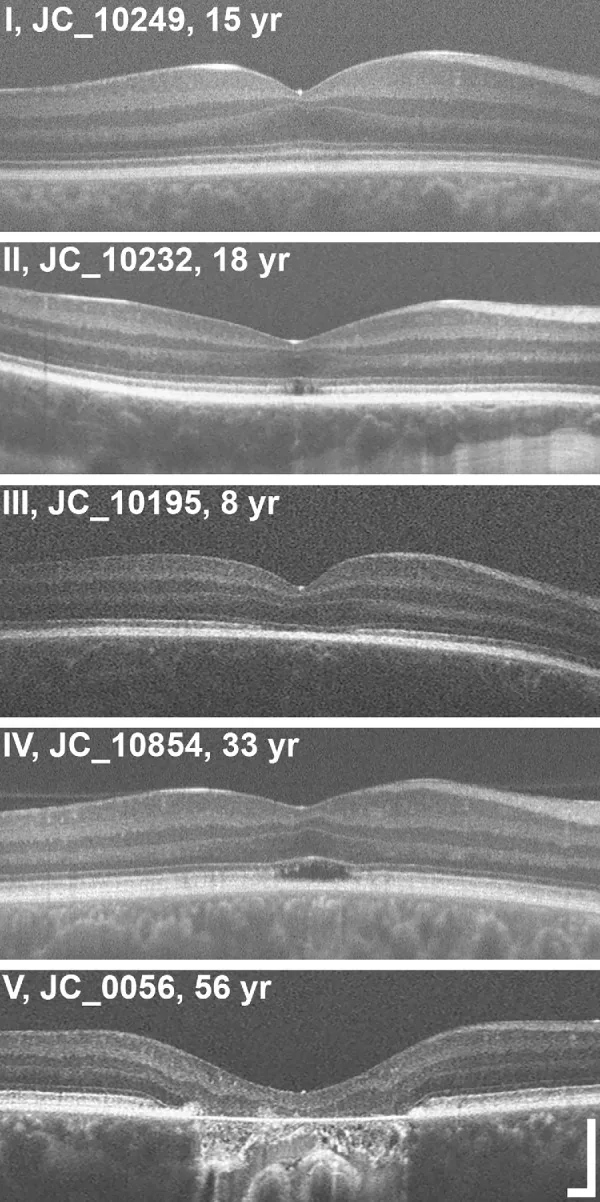
Grissim G, et al. Longitudinal Assessment of OCT-Based Measures of Foveal Cone Structure in Achromatopsia. Invest Ophthalmol Vis Sci. 2024. Figure 1. PMCID: PMC11005076. License: CC BY.
Fünfstufige Grad-Klassifikation der EZ im OCT: normale EZ (I), Unterbrechung der EZ (II), Verschwinden der EZ (III), Zone niedriger Reflektivität (IV), Atrophie der äußeren Netzhaut und RPE (V). Dies entspricht den Anomalien der Ellipsoidzone, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt werden.
Die Symptome der ACHM beginnen innerhalb der ersten Lebenswochen aufzutreten.
Verminderte Sehschärfe: Bei der vollständigen Form unter 20/200 (etwa 0,1), bei der unvollständigen Form etwa 20/80 (etwa 0,25).
Photophobie (Lichtempfindlichkeit): 38 % der befragten Patienten nannten sie als das wichtigste Symptom2). In heller Umgebung nimmt die Sehfunktion deutlich ab.
Hemeralopie (Tagblindheit): Die Sehfunktion ist bei hellem Licht vermindert, während sie bei schwachem Licht relativ gut ist.
Achromatopsie/Farbsehschwäche: Verlust des Farbsehens auf allen drei Achsen1). Die Ishihara-Farbtafeln sind bis auf die Demonstrationstafel fast unlesbar. Der Panel-D-15-Test zeigt kreisförmige, schräge Verwechslungslinien.
Pendelnystagmus: Tritt innerhalb der ersten Lebenswochen auf. Tendenz zur Besserung mit zunehmendem Alter und Abnahme beim Nahsehen.
Begleitende Hyperopie: Hyperopie ist häufig, aber einige weisen ein breites Spektrum an Refraktionsfehlern einschließlich Myopie auf.
Paradoxe Pupillenreaktion: Initiale Pupillenverengung im Dunkeln, ein charakteristischer Befund. Wichtiger Hinweis für die Diagnose bei Kindern.
Fundusbefunde: Anfangs fast normal. Die Fluoreszenzangiographie zeigt ebenfalls fast normale Befunde. Im Laufe der Zeit können fleckige Veränderungen und Atrophie des retinalen Pigmentepithels (RPE) auftreten. Häufig fehlt der Makulareflex und es liegt eine Makuladegeneration vor. Im OCT zeigen sich Unregelmäßigkeiten und Unschärfe der Ellipsoidzone (Verbindung von Innen- und Außensegment der Photorezeptoren) und der Interdigitationszone (Spitzen der Zapfenaußensegmente) in der äußeren Netzhautschicht.
Elektroretinographie-Befunde: Im photopischen ERG ist die Zapfenantwort stark reduziert oder fehlt, während die Stäbchenantwort im skotopischen ERG normal oder fast normal ist1)2). Beim GNAT2-Typ sind die S-Zapfen im Vergleich zu den CNGA3/CNGB3-Typen relativ besser erhalten.
FAF (Fundusautofluoreszenz): Es gibt vier Muster: normal, zentrales Signal erhöht, zentrales Signal vermindert, hyperfluoreszenter Ring + zentrale Hypofluoreszenz2).
OCT-Stadieneinteilung: Bewertung der strukturellen Veränderungen der äußeren Foveaschicht in fünf Stadien3).
Stadium
OCT-Befund
Stadium 1
Erhalt der äußeren Netzhaut (ELM-Hyperreflektivität, EZ-Abflachung)
Stadium 2
Zerstörung der Ellipsoidzone (EZ)
Stadium 3
Auftreten eines optisch leeren Raums
Stadium 4
Optisch leerer Raum + teilweise Zerstörung des RPE
Stadium 5
Verlust der äußeren Körnerschicht und/oder vollständige Zerstörung des RPE
In einer 10-Jahres-Follow-up-Studie wurde trotz stabiler bester korrigierter Sehschärfe (20/400 bis 20/200) eine strukturelle Progression im OCT bestätigt (Vergrößerung des optisch leeren Raums: rechtes Auge 246×59 μm, linkes Auge 326×53 μm)3).
Hyperreflektive Foci treten mehr als 3 Jahre vor Veränderungen der EZ auf und könnten ein früher Marker für das Fortschreiten der Erkrankung sein3).
AOSLO (Adaptive Optics Scanning Laser Ophthalmoskopie): Das Zapfenmosaik zeigt dunkle Räume, vergrößerte Zapfenabstände und eine verringerte Zapfendichte. Es gibt keinen signifikanten Unterschied zwischen CNGA3- und CNGB3-Typen, während der GNAT2-Typ relativ gut erhaltene Zapfen aufweist2).
Farbsehtest: Die Ishihara-Farbtafeln sind bis auf die Demonstrationsplatte nahezu unlesbar. Der Panel D-15 zeigt ein Fehlermuster entlang der skotopischen Achse (zwischen Deutan und Tritan). Das Anomaloskop zeigt eine steile Neigung und schließt den normalen Gleichgewichtsbereich nicht ein.
QVerschlechtert sich die Sehschärfe bei Achromatopsie mit dem Alter?
A
Die beste korrigierte Sehschärfe bleibt oft langfristig stabil. Andererseits kann die strukturelle Beurteilung mittels OCT eine Progression im Laufe der Zeit zeigen (Zerstörung der Ellipsoidzone, Vergrößerung des optisch leeren Raums)3). Eine Diskrepanz zwischen Funktion und Struktur ist charakteristisch, und eine regelmäßige Überwachung durch Untersuchungen ist wichtig.
Es handelt sich um einen autosomal-rezessiven Erbgang. Wenn beide Elternteile Träger sind, beträgt das Risiko für das Kind, die Erkrankung zu entwickeln, 25 %1). Die Erkrankung tritt meist bei Kindern von äußerlich gesunden Eltern auf, und eine Familienanamnese fehlt oft.
Auch ein nicht-mendelscher Vererbungsmodus durch väterliche uniparentale Disomie (UPD) wurde berichtet. In einem Fall, bei dem ein Homozygot für CNGA3 c.778G>C (p.D260H) durch UPD entstanden war, wurde bei der Mutter keine Mutation nachgewiesen 6). Solche Beispiele sind für die Risikobewertung des Wiederholungsrisikos in der genetischen Beratung von Bedeutung.
ATF6 (1q23.3): Transkriptionsfaktor der endoplasmatischen Retikulum-Stressantwort. Etwa 2%. Mechanismus, der nicht direkt an der Phototransduktion beteiligt ist1)2)
Niedrig penetranter Allel: CNGB3 c.1208G>A (p.R403Q) behält partielle Funktion, was zu einem milden Phänotyp führt2).
Merkmale von PDE6C-Mutationen: Bei 4 Fällen mit neuen Mutationen (c.1670G>A, c.2192G>A) wurde die Trias Nystagmus, Photophobie und Farbsehstörung bei allen festgestellt. Das Elektroretinogramm zeigte ein Verschwinden des photopischen und 30-Hz-Flicker-Signals bei normalem skotopischen Signal, und compound-heterozygote Patienten zeigten einen schwereren Phänotyp5).
Digenische Vererbung: Es gibt seltene Fälle mit Mutationen sowohl in CNGA3 als auch in CNGB31).
QWarum kann ein Kind erkranken, auch wenn beide Eltern nicht achromat sind?
A
Da die Achromatopsie autosomal-rezessiv vererbt wird, sind die Eltern, auch wenn sie jeweils eine Kopie des mutierten Gens tragen (Konduktoren), äußerlich gesund und haben normales Farbsehen. Bei einem Paar von Konduktoren besteht eine 25%ige Wahrscheinlichkeit, dass das Kind zwei Kopien der Mutation erbt und erkrankt1).
Bei Nystagmus, Photophobie und verminderter Sehschärfe innerhalb der ersten Lebenswochen ist eine multidisziplinäre Untersuchung unter Berücksichtigung einer ACHM erforderlich. Die definitive Diagnose erfordert einen Gentest.
Elektroretinogramm
Diagnostische Bedeutung: Goldstandard
Befunde der kompletten Form: Die Zapfenantwort im photopischen Elektroretinogramm fehlt oder ist stark reduziert. Die Stäbchenantwort im skotopischen Elektroretinogramm ist normal bis nahezu normal1)2)
Befunde der inkompletten Form: Eine schwache Zapfenantwort entsprechend der verbliebenen Zapfenfunktion wird nachgewiesen
Merkmale des GNAT2-Typs: S-Zapfen sind relativ besser erhalten als beim CNGA3/CNGB3-Typ
OCT
Diagnostische Bedeutung: Standard für die strukturelle Beurteilung
Bewertungsinhalt: 5-stufiges Staging der äußeren Schichten der Fovea, Nachweis einer Foveahypoplasie2)3)
Verlaufskontrolle: Auch bei funktioneller Stabilität kann ein strukturelles Fortschreiten auftreten, daher ist eine regelmäßige Überwachung wichtig
Zusatzuntersuchungen: Multimodale Beurteilung durch Kombination von FAF und AOSLO
Gentest
Diagnostische Bedeutung: Unverzichtbar für die definitive Diagnose
Methode: Gezieltes Panel von 6 Genen (CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)2)
Aufklärungsrate: Identifizierung des ursächlichen Gens in über 90% der Fälle möglich1)
Teilnahme an klinischen Studien: Eine molekulare Diagnose ist für die Teilnahme an klinischen Gentherapiestudien obligatorisch
Hyperopie ist häufig, aber es können verschiedene Refraktionsfehler auftreten. Die Korrektur mit Brille oder Kontaktlinsen ist wichtig, um die Sehschärfe zu maximieren. Bei begleitender Amblyopie sind Okklusion oder Atropintherapie zu erwägen.
Getönte Gläser zur Reduzierung der Photophobie verbessern die Lebensqualität erheblich. In einer Patientenbefragung bevorzugten 96 % Graufilter gegenüber Rotfiltern, und im Freien bevorzugten 74 % Graufilter2). Die Auswahl sollte an die Vorlieben und die Aktivitätsumgebung des einzelnen Patienten angepasst werden.
Eine Erklärung und Unterstützung durch Fachleute zu den Merkmalen des autosomal-rezessiven Erbgangs, dem Wiederholungsrisiko und der Bedeutung der genetischen Diagnostik ist erforderlich. Es wird auch empfohlen, sich einer genetischen Diagnostik zu unterziehen, um auf zukünftige klinische Studien zur Gentherapie vorbereitet zu sein.
QWelche Farbe von Lichtschutzbrillen ist geeignet?
A
In einer Patientenbefragung bevorzugten 96 % Graufilter gegenüber Rotfiltern, und im Freien bevorzugten 74 % Graufilter 2). Da es jedoch individuelle Unterschiede gibt, ist es wünschenswert, verschiedene Filter auszuprobieren und dann in Absprache mit einem Augenarzt oder Low-Vision-Spezialisten den für die eigene Aktivitätsumgebung geeigneten Filter auszuwählen.
Die normale Phototransduktionskaskade in Zapfenphotorezeptoren ist wie folgt 1).
Dunkelheit: Bei hoher intrazellulärer cGMP-Konzentration sind die CNG-Kanäle geöffnet, Na⁺ und Ca²⁺ strömen ein, die Zelle bleibt depolarisiert und setzt kontinuierlich Glutamat frei.
Lichteinwirkung: Opsin (Sehpigment) wird aktiviert → Transducin (G-Protein) wird aktiviert → Phosphodiesterase (PDE) wird aktiviert → cGMP wird abgebaut → CNG-Kanäle schließen → Hyperpolarisation → Hemmung der Glutamatfreisetzung.
Negative Rückkopplung: GCAP (Guanylatzyklase-aktivierendes Protein) bindet Ca²⁺ und hemmt die Aktivität der retGC, wodurch die cGMP-Produktion reguliert wird.
CNGA3-Mutation: Missense-Mutationen beeinträchtigen die Proteinfaltung, den intrazellulären Transport und die Membranintegration 1)7). Die S4-Transmembrandomäne ist eine Mutations-Hotspot. Über 150 Missense-Mutationen wurden berichtet, bei 103 war die Pathogenität unbestimmt, aber 3D-Struktur-Funktions-Analysen deuten darauf hin, dass 86,4 % ähnliche funktionelle Konsequenzen wie bekannte pathogene Mutationen haben 7).
CNGB3-Mutation: Nonsense- und Frameshift-Mutationen erzeugen verkürzte oder funktionslose Kanalproteine 1). Bei CNGB3-Mangel bleiben CNGA3-Homomere-Kanäle erhalten, sodass möglicherweise eine geringe Zapfenfunktion erhalten bleibt.
ATF6-Mutation: Transkriptionsfaktor, der an der ungefalteten Proteinantwort (UPR) des endoplasmatischen Retikulums beteiligt ist, jedoch nicht direkt in die Phototransduktionskaskade eingreift 1)2). Der Pathomechanismus unterscheidet sich von anderen Genen und wird noch erforscht.
Der CNG-Kanal hat eine tetramere Struktur (CNGA3 × 3 und CNGB3 × 1, nach einigen Berichten 2:2), jede Untereinheit besitzt sechs Transmembrandomänen, eine zyklische Nukleotidbindungsdomäne, eine C-Linker-Region und eine porenbildende Domäne 1).
Mechanismus der Zapfendegeneration und Progression
Die postnatale Entwicklung und Morphologie der Zapfen ist nahezu normal, und die Degeneration beginnt vermutlich im jungen Erwachsenenalter. Die Akkumulation von cGMP ist am degenerativen Prozess beteiligt, und in Tiermodellen wurde eine schnellere Progression in S-Zapfen-reichen Regionen gezeigt 1).
Traditionell galt ACHM als nicht-progressive Erkrankung. Langzeitbeobachtungen mittels OCT haben jedoch gezeigt, dass trotz nahezu stabiler bestkorrigierter Sehschärfe strukturelle Veränderungen (EZ-Unterbrechung, Erweiterung optischer Lücken) fortschreiten 3). Diese Funktions-Struktur-Dissoziation ist für die Bewertung des therapeutischen Fensters der Gentherapie von Bedeutung.
In der stäbchenfreien Zone der Fovea (dicht mit Zapfen besiedelter Bereich ohne Stäbchen) ist der Zapfenverlust ausgeprägt, während in der parafovealen Region Stäbchen zum EZ-Band beitragen und die Funktion kompensieren 3).
7. Aktuelle Forschung und Zukunftsperspektiven (Berichte aus der Forschungsphase)
Die Studie NCT03001310 (AAV8-hCARp.hCNGB3) mit 11 Erwachsenen und 12 Kindern (insgesamt 23) zeigte eine akzeptable Sicherheit. Eine Verbesserung des Farbsehens wurde bei 6/23, eine Verbesserung der Photophobie bei 11/20 und eine Verbesserung der Lebensqualität (QoL) bei 21/23 festgestellt. Bei hohen Dosen wurde auch eine Tendenz zu erhöhter intraokularer Entzündung beobachtet2).
In der AAV8.CNGA3 (RD-CURE)-Studie erhielten 9 Patienten 3 Dosen (1×10¹⁰ bis 1×10¹¹ vg/Auge). Die 1- und 3-Jahres-Daten zeigten einen Trend zur Verbesserung der Sehschärfe und des Kontrastsehens, erreichten jedoch keine statistische Signifikanz. Die Sicherheit war gut1)2).
McKyton et al. (2021) führten nach subretinaler Injektion von AAV2tYF-PR1.7-hCNGA3 (NCT02935517) bei zwei Erwachsenen mit CNGA3-ACHM eine fMRT-basierte kortikale visuelle Kartierung durch4). Das behandelte Auge zeigte im Vergleich zum unbehandelten Auge eine 5-fache Lichttoleranz (dramatische Verbesserung der Photophobie), den Erwerb von Rotdetektionsfähigkeit und eine Verkleinerung der Population Receptive Field (pRF)-Größe (was auf eine verbesserte räumliche Auflösung hindeutet). Andererseits wurde keine Aktivierung farbspezifischer kortikaler Areale (wie V4) festgestellt, und das Ganzfeld-Elektroretinogramm konnte keine Zapfenantwort nachweisen. Die Patienten berichteten über Verbesserungen im täglichen Leben, wie „erhöhtes Sicherheitsgefühl an Fußgängerüberwegen“, „keine Lupe mehr nötig“ und „draußen keine Sonnenbrille mehr nötig“.
In mehreren Tiermodellen wurde eine funktionelle Erholung nach Genersatz bestätigt1)2).
Mausmodell: Erholung auf 80% des Normalwerts im Elektroretinogramm.
Hundemodell (CNGB3): Erholung des Zapfen-Flicker-Elektroretinogramms nach 2,5 Jahren Nachbeobachtung nach Genersatz. Verhaltensverbesserung bei Umgebungen mit 25 Lux oder mehr.
Schafmodell (CNGA3): Langzeitverbesserung über mindestens 6 Jahre nach Genersatz.
Bezüglich des therapeutischen Zeitfensters haben Tierversuche gezeigt, dass eine Behandlung in jungen Jahren wirksamer ist. Alte Mäuse zeigten eine schlechte Reaktion, aber eine Vorbehandlung mit CNTF (ciliary neurotrophic factor) ermöglichte auch bei alten Hunden eine funktionelle Erholung1)2).
Eine klinische Studie (NCT04041232) mit Phenylbutyratglycerol (PBA), das auf die Stressreaktion des endoplasmatischen Retikulums abzielt, ist im Gange2). Dieser Ansatz zielt darauf ab, die Fehlfaltung des endoplasmatischen Retikulums zu mildern, anstatt das Gen zu ersetzen.
Die Hauptherausforderungen der Gentherapie sind folgende1)2)4).
Immunogenität: Die Immunantwort auf das AAV-Kapsid kann die therapeutische Wirksamkeit einschränken.
Dosisoptimierung: Notwendigkeit, das Gleichgewicht zwischen therapeutischer Wirkung und Risiko einer intraokularen Entzündung zu finden.
Entwicklungsbedingte Herausforderungen: Schwierigkeit der Operation bei Kindern und Behandlung von Amblyopie.
Grenzen der Plastizität des visuellen Kortex: Bei Erwachsenen kann eine kortikale Reorganisation schwierig sein, daher ist der Zeitpunkt der Behandlung entscheidend.
Langzeitwirksamkeit und Kosten: Sicherstellung einer dauerhaften Wirkung und medizinökonomische Herausforderungen.
QKann die Gentherapie die Achromatopsie vollständig heilen?
A
Derzeit befindet man sich in der Phase I/II der Sicherheits- und Wirksamkeitsstudien. Eine Verringerung der Photophobie und eine gewisse Verbesserung der Lichtempfindlichkeit wurden berichtet, aber eine vollständige Wiederherstellung des Farbensehens wurde nicht erreicht2)4). Tierversuche deuten darauf hin, dass eine Behandlung in jungen Jahren wirksam sein könnte, aber Langzeitdaten beim Menschen sind noch begrenzt. Es ist wichtig, die Fortschritte der Forschung zu verfolgen und regelmäßig Informationen mit dem behandelnden Arzt auszutauschen.
McKyton A, Averbukh E, Marks Ohana D, Levin N, Banin E. Cortical Visual Mapping following Ocular Gene Augmentation Therapy for Achromatopsia. J Neurosci. 2021;41(35):7363-7371. doi:10.1523/JNEUROSCI.3222-20.2021. PMID:34349002; PMCID:PMC8412991.
Madeira C, Godinho G, Grangeia A, Falcão M, Silva R, Carneiro Â, et al. Two Novel Disease-Causing Variants in the PDE6C Gene Underlying Achromatopsia. Case reports in ophthalmology. 2021;12(3):749-760. doi:10.1159/000512284. PMID:34720973; PMCID:PMC8460892.
Kohl S, Baumann B, Dassie F, et al. Paternal Uniparental Isodisomy of Chromosome 2 in a Patient with CNGA3-Associated Autosomal Recessive Achromatopsia. Int J Mol Sci. 2021;22(15):7842. doi:10.3390/ijms22157842. PMID:34360608; PMCID:PMC8346044.
Rasmussen DK, Sun YJ, Franco JA, et al. Structure-function analysis of CNGA3-associated achromatopsia patient variants complements clinical genomics in pathogenicity determination. Orphanet J Rare Dis. 2025;20:261. doi:10.1186/s13023-025-03792-3.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.