ภาวะตาบอดสีโดยสมบูรณ์ (Achromatopsia) เป็นโรคทางพันธุกรรมแบบถอย autosomal ที่พบได้ยาก โดยส่งผลต่อการทำงานของเซลล์รูปกรวย ทั้งสามชนิด โดยมีความชุกประมาณ 1 ใน 30,000 คนทั่วโลก
อาการหลัก ได้แก่ การมองเห็น ลดลง กลัวแสง (ไวต่อแสง ) อาตา ไม่สามารถมองเห็นสี และตาบอดกลางวัน โดยอาการจะปรากฏภายในไม่กี่สัปดาห์หลังคลอด
ยีนที่เป็นสาเหตุมี 6 ชนิด ได้แก่ CNGA3, CNGB3, GNAT2, PDE6C, PDE6H และ ATF6 โดย CNGA3 และ CNGB3 คิดเป็น 80-90% ของผู้ป่วยทั้งหมด
ปัจจุบันยังไม่มีการรักษาที่หายขาดที่ได้รับการอนุมัติ การรักษาจึงเน้นตามอาการด้วยแว่นตากันแสง การแก้ไขสายตาผิดปกติ และการดูแลผู้ที่มีสายตาเลือนราง
ค่าสายตาที่ดีที่สุดที่แก้ไขแล้ว (BCVA) โดยทั่วไปจะคงที่ในระยะยาว แต่การเปลี่ยนแปลงโครงสร้างจากการตรวจ OCT จะแสดงความคืบหน้าเมื่อเวลาผ่านไป
มีการทดลองทางคลินิกเกี่ยวกับการบำบัดด้วยยีน (โดยใช้พาหะ AAV) หลายรายการที่กำลังดำเนินอยู่ โดยมีรายงานว่าสามารถลดอาการกลัวแสง และปรับปรุงการทำงานของการมองเห็น บางส่วน แต่ยังไม่สามารถฟื้นฟูการมองเห็น สีได้อย่างสมบูรณ์
ภาวะตาบอดสีโดยสมบูรณ์ (Achromatopsia; ACHM) เป็นโรคจอประสาทตา ทางพันธุกรรมที่พบได้ยากในตาทั้งสองข้าง โดยเซลล์รูปกรวย ทั้งสามชนิดสูญเสียการทำงาน โรคนี้ยังเรียกว่า “rod monochromatism” หรือ “total color blindness” 1)
ความชุกประมาณ 1 ใน 30,000 คนทั่วโลก 1) โรคนี้ถ่ายทอดทางพันธุกรรมแบบถ่ายทอดทาง autosomal recessive ไม่มีความผิดปกติของระบบอื่นร่วมด้วย และอายุขัยเฉลี่ยปกติ
ACHM มีสองประเภท: สมบูรณ์ และ ไม่สมบูรณ์ ในประเภทสมบูรณ์ การทำงานของเซลล์รูปกรวย ทั้งหมดจะหายไป ส่วนประเภทไม่สมบูรณ์ เซลล์รูปกรวย อย่างน้อยหนึ่งชนิดยังคงทำงานได้บ้าง โดยค่าสายตาประมาณ 20/40 ถึง 20/120 และอาการกลัวแสง และอาตา จะน้อยกว่า 2)
ยีนที่เป็นสาเหตุมี 6 ชนิด (CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, ATF6) และสามารถระบุยีนที่เป็นสาเหตุได้ในมากกว่า 90% ของผู้ป่วย 1) 2) โดยยีน CNGA3 และ CNGB3 เพียงสองชนิดคิดเป็น 80-90% ของผู้ป่วยทั้งหมด
โปรดทราบว่าความผิดปกติของการมองเห็นสี ทั่วไป (ความผิดปกติของการมองเห็นสี แต่กำเนิด) เกิดจากความผิดปกติของเม็ดสีรับภาพในเซลล์รูปกรวย หนึ่งหรือสองชนิด และส่งผลต่อการมองเห็น สีเท่านั้น ACHM แตกต่างโดยพื้นฐานเนื่องจากเซลล์รูปกรวย ทั้งหมดไม่ทำงาน ร่วมกับการมองเห็น ลดลง อาตา และกลัวแสง
ภาวะตาบอดสีเซลล์รูปกรวย สีน้ำเงิน (BCM) เป็นการถ่ายทอดทางพันธุกรรมแบบเชื่อมโยงกับโครโมโซม X โดยที่เซลล์รูปแท่ง และเซลล์รูปกรวย S (สีน้ำเงิน) ทำงาน แต่ขาดเซลล์รูปกรวย L และ M การมองเห็น 0.1-0.3 มีอาการคล้าย ACHM แต่รูปแบบการถ่ายทอดทางพันธุกรรมและการทำงานของเซลล์รูปกรวย ที่เหลือแตกต่างกัน ดังนั้นการวินิจฉัยแยกโรคจึงมีความสำคัญในการกำหนดกลยุทธ์การรักษา
ภาวะตาบอดสีจากสมอง เป็นความผิดปกติของการมองเห็นสี ที่เกิดขึ้นภายหลังเนื่องจากรอยโรคในบริเวณการมองเห็น V4 ของสมอง (เช่น โรคหลอดเลือดสมอง เนื้องอก) และเป็นโรคที่แตกต่างอย่างสิ้นเชิงจาก ACHM ที่จอประสาทตา แต่กำเนิด
มีกรณีที่รู้จักกันดีคือ ผลกระทบจากผู้ก่อตั้งบนเกาะปิงเกลาป (ไมโครนีเซีย) หลังจากพายุไต้ฝุ่นในคริสต์ศตวรรษที่ 1700 ประชากรบนเกาะลดลงอย่างมาก และการกลายพันธุ์ CNGB3 (p.S435F) แพร่กระจายในหมู่ผู้รอดชีวิต ทำให้มีความชุกประมาณ 10% และอัตราการเป็นพาหะประมาณ 30%1) 2)
Q
ภาวะตาบอดสีทั้งหมดกับความผิดปกติของการมองเห็นสีทั่วไป (ตาบอดสีบางส่วน) แตกต่างกันอย่างไร?
A
ความผิดปกติของการมองเห็นสี แต่กำเนิดทั่วไปเกิดจากความผิดปกติของเม็ดสีรับภาพในเซลล์รูปกรวย หนึ่งหรือสองชนิด และส่งผลต่อการมองเห็น สีเท่านั้น โดยการมองเห็น อยู่ในเกณฑ์ปกติ ในภาวะตาบอดสีทั้งหมด การทำงานของเซลล์รูปกรวย ทั้งสามชนิดบกพร่อง ส่งผลให้สูญเสียการมองเห็น สี ร่วมกับการมองเห็น ลดลง (ประมาณ 0.1 หรือน้อยกว่า) อาตา กลัวแสง และตาบอดกลางวัน ซึ่งแตกต่างกันโดยพื้นฐาน
Grissim G, et al. Longitudinal Assessment of
OCT -Based Measures of Foveal Cone Structure in Achromatopsia. Invest Ophthalmol Vis Sci. 2024. Figure 1. PM
CI D: PMC11005076. License: CC BY.
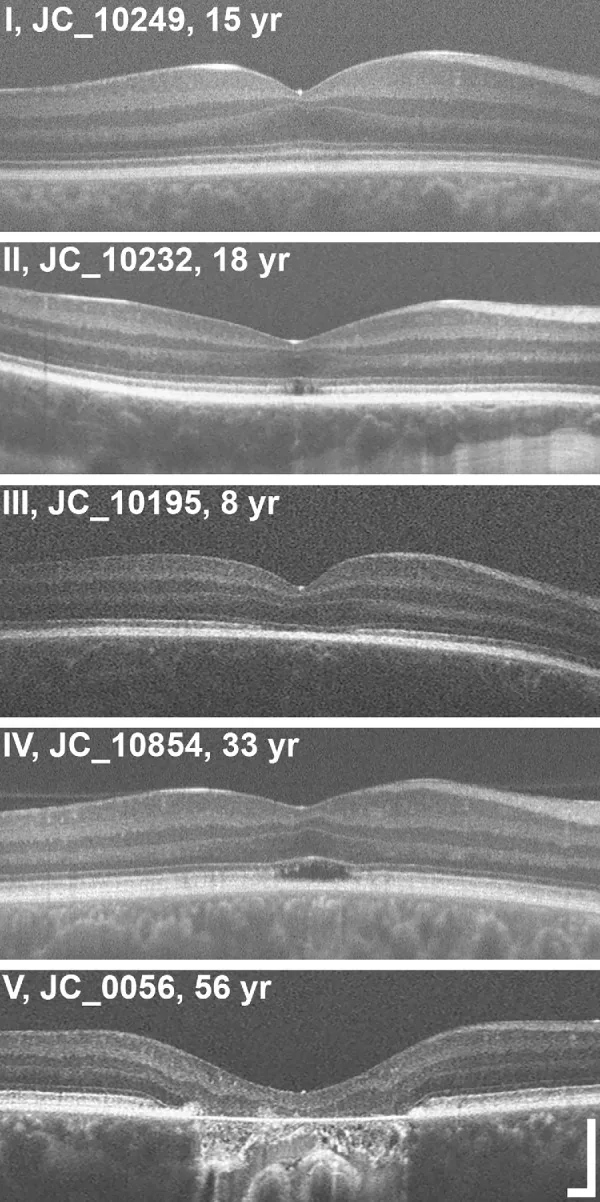
การจำแนกระดับ EZ ห้าระดับโดย OCT แสดง EZ ปกติ (I), การขาดตอนของ EZ (II), การหายไปของ EZ (III), บริเวณสะท้อนแสงต่ำ (IV) และการฝ่อของจอประสาทตา ชั้นนอกและ RPE (V) สอดคล้องกับความผิดปกติของบริเวณ ellipsoid ที่กล่าวถึงในหัวข้อ “2. อาการหลักและผลการตรวจทางคลินิก”
อาการของ ACHM เริ่มปรากฏภายในไม่กี่สัปดาห์แรกหลังคลอด
การมองเห็น ลดลงกลัวแสง (ไวต่อแสง ) : จากการสำรวจผู้ป่วย 38% รายงานว่าเป็นอาการที่สำคัญที่สุด2) การทำงานของการมองเห็น ลดลงอย่างมากในสภาพแวดล้อมที่สว่างตาบอดกลางวัน (hemeralopia) : การทำงานของการมองเห็น ลดลงในที่สว่าง แต่ค่อนข้างดีในสภาพแวดล้อมที่มืดการสูญเสีย/ลดลงของการมองเห็น สี : สูญเสียการมองเห็น สีในทั้งสามแกน1) ในการทดสอบอิชิฮาระ แผ่นทดสอบส่วนใหญ่ไม่สามารถอ่านได้ยกเว้นแผ่นสาธิต การทดสอบ Panel D-15 แสดงเส้นสับสนแบบวงกลมเฉียงอาตา แบบลูกตุ้มความสัมพันธ์กับสายตายาว : มักพบสายตายาว แต่อาจมีค่าสายตาผิดปกติหลายแบบรวมถึงสายตาสั้น
ปฏิกิริยารูม่านตา ที่ขัดแย้ง : การหดตัวของรูม่านตา เริ่มแรกในที่มืด เป็นลักษณะเด่นที่สำคัญในการวินิจฉัยเด็ก
ผลการตรวจอวัยวะภายในตา : ระยะแรกดูเกือบปกติ การถ่ายภาพหลอดเลือดด้วยฟลูออเรสซีน ก็แสดงผลเกือบปกติเช่นกัน เมื่อเวลาผ่านไป อาจเกิดการเปลี่ยนแปลงแบบจุดด่างและการฝ่อของเยื่อบุผิวเม็ดสีจอประสาทตา มักพบการหายไปของรีเฟล็กซ์โฟเวียหรือจอประสาทตา เสื่อม ในการตรวจ OCT รอยต่อระหว่างส่วนในและส่วนนอกของเซลล์รับแสง และโซนอินเตอร์ดิจิเทชันไม่สม่ำเสมอและไม่ชัดเจน
ผลการตรวจคลื่นไฟฟ้าจอประสาทตา : ใน ERG แบบสว่าง การตอบสนองของเซลล์รูปกรวย ลดลงอย่างมากหรือหายไป ในขณะที่การตอบสนองของเซลล์รูปแท่ง ใน ERG แบบมืดเป็นปกติหรือเกือบปกติ1) 2) ในชนิด GNAT2 เซลล์รูปกรวย S ถูกเก็บรักษาไว้ค่อนข้างดีกว่าชนิด CNGA3/CNGB3
ภาวะโฟเวียเจริญไม่เต็มที่ : พบใน 60-70% ของกรณีที่มีการกลายพันธุ์ CNGA3/CNGB32)
FAF (การเรืองแสงเองของจอประสาทตา )2)
การแบ่งระยะ OCT : ประเมินการเปลี่ยนแปลงโครงสร้างของชั้นนอกของโฟเวียเป็น 5 ระยะ3)
ระยะ ผล OCT ระยะที่ 1 การคงไว้ของชั้นนอกจอประสาทตา (ELM สะท้อนแสงมาก, EZ แบนราบ) ระยะที่ 2 การทำลายโซน ellipsoid (EZ ) ระยะที่ 3 การปรากฏของช่องว่างเชิงแสง (optically empty space) ระยะที่ 4 ช่องว่างเชิงแสง + การทำลาย RPE บางส่วน ระยะที่ 5 การหายไปของชั้นนิวเคลียสชั้นนอก และ/หรือ การทำลาย RPE อย่างสมบูรณ์
ในการศึกษาติดตามผล 10 ปี แม้ว่าค่าความคมชัดของการมองเห็น ที่ดีที่สุดที่แก้ไขแล้วจะคงที่ (20/400 ถึง 20/200) แต่การดำเนินไปทางโครงสร้างใน OCT (การขยายตัวของช่องว่างเชิงแสง: ตาขวา 246×59 μm, ตาซ้าย 326×53 μm) ได้รับการยืนยัน3) .
จุดสะท้อนแสงสูง (hyperreflective foci) อาจปรากฏขึ้นก่อนการเปลี่ยนแปลงของ EZ มากกว่า 3 ปี ซึ่งบ่งชี้ถึงศักยภาพในการเป็นเครื่องหมายบ่งชี้ระยะเริ่มต้นของการดำเนินโรค3) .
AO SLO (กล้องตรวจตาเลเซอร์สแกนแบบปรับแสงได้)เซลล์รูปกรวย แสดงพื้นที่มืด (dark space) ระยะห่างระหว่างเซลล์รูปกรวย เพิ่มขึ้น และความหนาแน่นของเซลล์รูปกรวย ลดลง ไม่มีความแตกต่างอย่างมีนัยสำคัญระหว่างชนิด CNGA3 และ CNGB3 ในขณะที่ชนิด GNAT2 ค่อนข้างคงสภาพเซลล์รูปกรวย ไว้2) .
การทดสอบการมองเห็นสี
Q
การมองเห็นในโรคตาบอดสีสมบูรณ์แย่ลงตามอายุหรือไม่?
A
ค่าความคมชัดของการมองเห็น ที่ดีที่สุดที่แก้ไขแล้วมักจะคงที่ในระยะยาว ในทางกลับกัน การประเมินโครงสร้างด้วย OCT อาจแสดงการเปลี่ยนแปลงเมื่อเวลาผ่านไป (การทำลายโซนรูปไข่ การขยายตัวของช่องว่างเชิงแสง) ซึ่งอาจดำเนินไป3) มีการแยกกันระหว่างการทำงานและโครงสร้าง และการติดตามอย่างสม่ำเสมอด้วยการตรวจเป็นระยะเป็นสิ่งสำคัญ
การถ่ายทอดแบบ autosomal recessive หากทั้งพ่อและแม่เป็นพาหะ (ผู้携带基因) ความเสี่ยงที่บุตรจะเป็นโรคคือ 25%1) มักเกิดในบุตรของพ่อแม่ที่ดูแข็งแรงดี และในหลายกรณีไม่มีประวัติครอบครัว
นอกจากนี้ยังมีรายงานรูปแบบการถ่ายทอดทางพันธุกรรมแบบไม่เป็นเมนเดเลียนเนื่องจากภาวะยูนิพาเรนทัลไดโซมี (UPD) จากฝ่ายบิดา ในกรณีที่พบโฮโมไซกัสของ CNGA3 c.778G>C (p.D260H) ซึ่งเกิดจาก UPD ไม่พบการกลายพันธุ์ในมารดา 6) ตัวอย่างดังกล่าวมีความสำคัญอย่างยิ่งในการประเมินความเสี่ยงของการเกิดซ้ำในการให้คำปรึกษาทางพันธุกรรม
CNGA3
โครโมโซม : 2q11.2
หน้าที่ : หน่วยย่อยอัลฟาของช่อง CNG
ความถี่ : ประมาณ 25-50% ของผู้ป่วย1) 2)
รูปแบบการกลายพันธุ์ : การกลายพันธุ์แบบมิสเซนส์เป็นหลัก โดเมนทรานส์เมมเบรน S4 เป็นจุดร้อน
การกระจายทางภูมิศาสตร์ : ในตะวันออกกลางและจีน CNGA3 คิดเป็นมากกว่า 80%
CNGB3
โครโมโซม : 8q21.3
หน้าที่ : หน่วยย่อยเบตาของช่อง CNG
ความถี่ : ประมาณ 50% ของผู้ป่วย1) 2)
รูปแบบการกลายพันธุ์ : การกลายพันธุ์แบบนอนเซนส์, เฟรมชิฟต์ และสไปซิงเป็นหลัก c.1148delC เป็นการกลายพันธุ์ที่พบบ่อยที่สุด
การกระจายทางภูมิศาสตร์ : ในยุโรปและอเมริกา CNGB3 คิดเป็นมากกว่า 50%
ยีนอื่นๆ
GNAT2 (1p13.3): ทรานส์ดิวซินอัลฟาของเซลล์รูปกรวย ประมาณ 2% ค่อนข้างไม่รุนแรง โดยชั้นเซลล์รับแสง ยังคงอยู่ 2)
PDE6C (10q23.33): หน่วยย่อยอัลฟาของ PDE ในเซลล์รูปกรวย ชนิดรุนแรงที่เริ่มมีอาการเร็ว 5)
PDE6H (12p12.3): หน่วยย่อยแกมมาของ PDE ในเซลล์รูปกรวย พบได้น้อยมาก 1)
ATF6 (1q23.3): ปัจจัยถอดรหัสตอบสนองต่อความเครียดของเอนโดพลาสมิกเรติคูลัม ประมาณ 2% กลไกที่ไม่เกี่ยวข้องโดยตรงกับการส่งผ่านแสง 1) 2)
อัลลีลที่มีการแทรกซึมต่ำ : CNGB3 c.1208G>A (p.R403Q) ยังคงการทำงานบางส่วน ส่งผลให้มีฟีโนไทป์ไม่รุนแรง 2)
ลักษณะของการกลายพันธุ์ PDE6C : ใน 4 รายที่มีการกลายพันธุ์ใหม่ (c.1670G>A, c.2192G>A) พบสามอาการ (ตากระตุก กลัวแสง ความผิดปกติของการมองเห็นสี ) ในทุกราย การตรวจคลื่นไฟฟ้าจอประสาทตา แสดงให้เห็นว่าการมองเห็น ในที่สว่างและการกะพริบ 30 Hz หายไป ในขณะที่การมองเห็น ในที่มืดปกติ และผู้ที่มีเฮเทอโรไซกัสเชิงซ้อนมีฟีโนไทป์ที่รุนแรงกว่า 5)
การถ่ายทอดทางพันธุกรรมแบบสองยีน : มีรายงานผู้ป่วยที่หายากซึ่งมีการกลายพันธุ์ทั้งใน CNGA3 และ CNGB3 1)
การให้คำปรึกษาทางพันธุกรรม
ตาบอดสีสมบูรณ์เป็นโรคถ่ายทอดทางพันธุกรรมแบบออโตโซมัลด้อย ผู้ป่วยมีการกลายพันธุ์สองชุด แต่บุตรของผู้ป่วยจะเกิดโรคได้ก็ต่อเมื่อคู่สมรสเป็นพาหะของยีนเดียวกัน (ความน่าจะเป็น 25%) การตรวจวินิจฉัยทางพันธุกรรมช่วยให้ประเมินความเสี่ยงในครอบครัวได้ แนะนำให้ปรึกษาแพทย์พันธุศาสตร์หรือที่ปรึกษาทางพันธุกรรมที่ได้รับการรับรอง
Q
เหตุใดเด็กจึงเป็นตาบอดสีสมบูรณ์ได้แม้ว่าพ่อแม่ทั้งสองคนจะไม่เป็นโรค?
A
เนื่องจากตาบอดสีสมบูรณ์เป็นโรคถ่ายทอดทางพันธุกรรมแบบออโตโซมัลด้อย พ่อและแม่อาจเป็นพาหะมียีนกลายพันธุ์คนละหนึ่งชุด (พาหะ) โดยไม่มีอาการและมีการมองเห็น สีปกติ เมื่อพาหะสองคนมีบุตร ความน่าจะเป็นที่บุตรจะได้รับยีนกลายพันธุ์สองชุดและเกิดโรคคือ 25% 1)
หากพบอาการตากระตุก กลัวแสง และการมองเห็น ลดลงภายในไม่กี่สัปดาห์แรกหลังคลอด จำเป็นต้องตรวจอย่างครอบคลุมโดยคำนึงถึงความเป็นไปได้ของ ACHM การตรวจทางพันธุกรรมเป็นสิ่งจำเป็นสำหรับการวินิจฉัยที่แน่นอน
การตรวจคลื่นไฟฟ้าจอประสาทตา
ความสำคัญในการวินิจฉัย : มาตรฐานทองคำ
ผลการตรวจชนิดสมบูรณ์ : การตอบสนองของเซลล์รูปกรวย ในการตรวจคลื่นไฟฟ้าจอประสาทตา แบบสว่างหายไปหรือลดลงอย่างมาก การตอบสนองของเซลล์รูปแท่ง ในการตรวจคลื่นไฟฟ้าจอประสาทตา แบบมืดปกติถึงเกือบปกติ1) 2)
ผลการตรวจชนิดไม่สมบูรณ์ : ตรวจพบการตอบสนองของเซลล์รูปกรวย ที่อ่อนแอซึ่งสอดคล้องกับการทำงานของเซลล์รูปกรวย ที่เหลืออยู่
ลักษณะเฉพาะของชนิด GNAT2 : เซลล์รูปกรวย S ถูกเก็บรักษาไว้ค่อนข้างดีกว่าชนิด CNGA3/CNGB3
OCT
ความสำคัญในการวินิจฉัย : มาตรฐานสำหรับการประเมินโครงสร้าง
เนื้อหาการประเมิน : การแบ่งระยะ 5 ระดับของชั้นนอกของรอยบุ๋มจอตา การตรวจหาภาวะรอยบุ๋มจอตา พัฒนาน้อย2) 3)
การติดตามผล : แม้จะคงที่ในด้านการทำงาน แต่การดำเนินไปทางโครงสร้างอาจเกิดขึ้นได้ ดังนั้นการติดตามอย่างสม่ำเสมอจึงสำคัญ
การตรวจเสริม : การประเมินแบบหลายรูปแบบที่รวม FAF และ AO SLO
การตรวจทางพันธุกรรม
ความสำคัญในการวินิจฉัย : จำเป็นสำหรับการวินิจฉัยที่แน่นอน
วิธีการ : แผงยีนเป้าหมายสำหรับ 6 ยีน (CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)2)
อัตราการระบุ : สามารถระบุยีนก่อโรคได้ในมากกว่า 90% ของกรณี1)
การเข้าร่วมการทดลองทางคลินิก : การวินิจฉัยระดับโมเลกุลเป็นสิ่งจำเป็นสำหรับการเข้าร่วมการทดลองทางคลินิกของการบำบัดด้วยยีน
ตารางทดสอบการมองเห็น สีอิชิฮาระ : แทบจะอ่านไม่ได้ยกเว้นตารางสาธิตแผ่นสี D-15/100 Hue (Farnsworth-Munsell) : รูปแบบความผิดพลาดบนแกน scotopic (ระหว่าง deutan และ tritan)เครื่อง Anomaloscope (ชนิด Nagel) : แสดงความชันสูงและไม่ครอบคลุมช่วงสีสมดุลปกติ
สิ่งสำคัญคือต้องแยก ACHM ออกจากโรคที่มีลักษณะทางคลินิกคล้ายคลึงกัน2)
ตาบอดสีเซลล์รูปกรวย สีน้ำเงิน (BCM) : ถ่ายทอดทางพันธุกรรมแบบ X-linked เซลล์รูปกรวย S ยังทำงานอยู่ สามารถแยกได้ด้วยเครื่อง Anomaloscopeโรคจอประสาทตา เสื่อมชนิดเซลล์รูปกรวย : ดำเนินไปหลังจากเริ่มมีอาการ แยกโดยรูปแบบคลื่นไฟฟ้าจอตาและระยะดำเนินโรคกลุ่มอาการ Alström : จอประสาทตา เสื่อมทั้งเซลล์รูปกรวย และเซลล์รูปแท่ง ร่วมกับอาการทั่วร่างกาย (อ้วน สูญเสียการได้ยิน ฯลฯ)ตาบอดกลางคืนแต่กำเนิดชนิดคงที่ (CSNB )เซลล์รูปแท่ง เป็นหลัก อาการหลักคือตาบอดกลางคืน ไม่ใช่ตาบอดกลางวันตาบอดสีทั้งหมด จากสมอง
ปัจจุบันยังไม่มีการรักษาที่หายขาดซึ่งได้รับการรับรองสำหรับ ACHM1) 2) การรักษามุ่งเน้นไปที่การบรรเทาอาการ
แม้ว่าสายตายาว จะพบได้บ่อย แต่มีความผิดปกติของการหักเหแสงหลายประเภท ดังนั้นการแก้ไขด้วยแว่นตาหรือคอนแทคเลนส์จึงมีความสำคัญต่อการเพิ่มการมองเห็น ให้สูงสุด หากมีภาวะตาขี้เกียจ ให้พิจารณาการปิดตาหรือการใช้ยาอะโทรพีน
เลนส์ป้องกันแสงเพื่อลดอาการกลัวแสง ช่วยปรับปรุงคุณภาพชีวิตประจำวันได้อย่างมาก ข้อมูลจากการสำรวจผู้ป่วยพบว่า 96% ชอบฟิลเตอร์สีเทามากกว่าสีแดง และ 74% ชอบฟิลเตอร์สีเทาเมื่ออยู่กลางแจ้ง2) การเลือกให้เหมาะสมกับความชอบส่วนบุคคลและสภาพแวดล้อมกิจกรรมของผู้ป่วยเป็นสิ่งสำคัญ
มีการใช้อุปกรณ์ช่วยและเทคโนโลยีดังต่อไปนี้ 3) .
อุปกรณ์ช่วยทางแสง : แว่นขยาย, เครื่องขยายภาพดิจิทัลอุปกรณ์ช่วยทางอิเล็กทรอนิกส์ : อุปกรณ์สวมใส่, แอปพลิเคชันสมาร์ทโฟนเทคโนโลยีการเข้าถึง : การอ่านข้อความออกเสียง, ซอฟต์แวร์ขยายหน้าจอ, การช่วยเหลือทางไกล
จำเป็นต้องมีคำอธิบายและการสนับสนุนจากผู้เชี่ยวชาญเกี่ยวกับลักษณะการถ่ายทอดทางพันธุกรรมแบบด้อยบนออโตโซม ความเสี่ยงในการเกิดซ้ำ และความสำคัญของการวินิจฉัยทางพันธุกรรม นอกจากนี้ ควรเข้ารับการวินิจฉัยทางพันธุกรรมเพื่อเตรียมพร้อมสำหรับการเข้าร่วมการทดลองทางคลินิกของการบำบัดด้วยยีน ในอนาคต
ปัจจุบันยังไม่มีการรักษาที่หายขาด และการบำบัดด้วยยีน ยังอยู่ในขั้นตอนการทดลองทางคลินิก ควรระวังการรักษาที่ไม่ได้รับการรับรองซึ่งโฆษณาว่าเป็น “การบำบัดด้วยยีน ”
สีและความเข้มของเลนส์กรองแสงแตกต่างกันมากในแต่ละบุคคล ควรปรึกษาจักษุแพทย์หรือผู้เชี่ยวชาญด้านการดูแลผู้มีสายตาเลือนราง เพื่อปรับให้เหมาะสม
การติดตามโครงสร้างจอประสาทตา อย่างสม่ำเสมอด้วย OCT มีความสำคัญในการกำหนดเวลาที่เหมาะสมสำหรับการแทรกแซงการรักษาในอนาคต
Q
สีใดที่เหมาะสมสำหรับแว่นตากรองแสง?
A
จากการสำรวจผู้ป่วย 96% ชอบฟิลเตอร์สีเทามากกว่าฟิลเตอร์สีแดง และ 74% ชอบฟิลเตอร์สีเทาเมื่ออยู่กลางแจ้ง 2) อย่างไรก็ตาม เนื่องจากความแตกต่างระหว่างบุคคล ควรลองใช้ฟิลเตอร์หลายๆ แบบ และเลือกฟิลเตอร์ที่เหมาะสมกับสภาพแวดล้อมในกิจกรรมของคุณ โดยปรึกษาจักษุแพทย์หรือผู้เชี่ยวชาญด้านสายตาเลือนราง
ลำดับการส่งสัญญาณแสงในเซลล์รูปกรวย ปกติมีดังนี้1)
ในที่มืด : ความเข้มข้นของ cGMP ภายในเซลล์สูง ช่อง CNG เปิด Na⁺ และ Ca²⁺ ไหลเข้า เซลล์คงสภาพดีโพลาไรเซชัน และปล่อยกลูตาเมตอย่างต่อเนื่องเมื่อสัมผัสแสง : ออปซิน (รงควัตถุทางการมองเห็น ) ถูกกระตุ้น → ทรานส์ดิวซิน (โปรตีน G) ถูกกระตุ้น → ฟอสโฟไดเอสเทอเรส (PDE) ถูกกระตุ้น → cGMP ถูกย่อยสลาย → ช่อง CNG ปิด → ไฮเปอร์โพลาไรเซชัน → ยับยั้งการปล่อยกลูตาเมตการป้อนกลับเชิงลบ : GCAP (โปรตีนกระตุ้นกัวนิเลตไซเคลส) จับกับ Ca²⁺ และยับยั้งการทำงานของ retGC ควบคุมการผลิต cGMP
การกลายพันธุ์ CNGA3 : การกลายพันธุ์แบบมิสเซนส์รบกวนการพับโปรตีน การขนส่งภายในเซลล์ และการรวมตัวในเยื่อหุ้ม1) 7) โดเมนทรานส์เมมเบรน S4 เป็นจุดร้อนของการกลายพันธุ์ มีรายงานการกลายพันธุ์แบบมิสเซนส์มากกว่า 150 ชนิด โดย 103 ชนิดยังไม่ทราบความก่อโรค แต่การวิเคราะห์โครงสร้าง-หน้าที่ 3 มิติชี้ให้เห็นว่า 86.4% มีผลทางหน้าที่คล้ายกับการกลายพันธุ์ที่ก่อโรคที่ทราบแล้ว7)
การกลายพันธุ์ CNGB3 : การกลายพันธุ์แบบนอนเซนส์และเฟรมชิฟต์สร้างโปรตีนช่องที่ถูกตัดทอนหรือสูญเสียหน้าที่1) เมื่อขาด CNGB3 ช่องโฮโมเมอร์ CNGA3 ยังคงอยู่ ดังนั้นอาจคงการทำงานของเซลล์รูปกรวย ไว้ได้บ้าง
การกลายพันธุ์ ATF6 : เป็นปัจจัยถอดรหัสที่เกี่ยวข้องกับการตอบสนองต่อโปรตีนผิดรูป (UPR) ของเอนโดพลาสมิกเรติคูลัม และไม่มีส่วนร่วมโดยตรงในลำดับการส่งสัญญาณแสง1) 2) กลไกพยาธิสภาพแตกต่างจากยีนอื่น และยังอยู่ระหว่างการศึกษา
ช่อง CNG มีโครงสร้างเป็นเตตระเมอร์ (CNGA3 × 3 และ CNGB3 × 1 ในบางรายงานเป็น 2:2) แต่ละหน่วยย่อยมีโดเมนทรานส์เมมเบรน 6 โดเมน โดเมนจับนิวคลีโอไทด์แบบวง บริเวณเชื่อมต่อ C และโดเมนสร้างรูพรุน1)
การพัฒนาหลักของเซลล์รูปกรวย หลังคลอดและสัณฐานวิทยาใกล้เคียงปกติ และเชื่อว่าการเสื่อมเริ่มต้นในวัยผู้ใหญ่ตอนต้น การสะสมของ cGMP เกี่ยวข้องกับกระบวนการเสื่อม และการดำเนินโรคที่เร็วกว่าในบริเวณที่อุดมด้วยเซลล์รูปกรวย S ได้แสดงให้เห็นในแบบจำลองสัตว์1)
แต่เดิม ACHM ถือเป็นโรคที่ไม่ดำเนินต่อ อย่างไรก็ตาม การสังเกตระยะยาวด้วย OCT แสดงให้เห็นว่าแม้การมองเห็น ที่ดีที่สุดที่แก้ไขแล้วจะค่อนข้างคงที่ แต่การเปลี่ยนแปลงโครงสร้าง (การทำลาย EZ การขยายช่องว่างทางแสง) ยังคงดำเนินต่อไป3) การแยกกันระหว่างหน้าที่และโครงสร้างนี้มีความสำคัญในการประเมินหน้าต่างการรักษาสำหรับการบำบัดด้วยยีน
ในบริเวณที่ไม่มีเซลล์รูปแท่ง (บริเวณที่มีเซลล์รูปกรวย หนาแน่น) ของรอยบุ๋มจอตา การสูญเสียเซลล์รูปกรวย จะเด่นชัด ในขณะที่บริเวณรอบรอยบุ๋มจอตา เซลล์รูปแท่ง จะชดเชยการทำงานโดยมีส่วนร่วมในแถบ EZ 3)
ด้านล่างนี้คือการทดลองทางคลินิกหลักของการบำบัดด้วยยีน ที่กำลังดำเนินการหรือเสร็จสิ้น ณ ปี 2024 1) 2)
หมายเลข NCT ยีนเป้าหมาย พาหะ สถานะ NCT 03001310CNGB3 AAV8-hCARp.hCNGB3 เสร็จสิ้น NCT 02610582CNGA3 AAV8-hG1.7-hCNGA3 เสร็จสิ้น NCT 03758404CNGA3 rAAV8.hCNGA3 กำลังรับสมัคร NCT 02599922CNGB3 AAV2tYF-PR1.7-hCNGB3 (AGTC-401) กำลังดำเนินการ NCT 02935517CNGA3 AAV2tYF-PR1.7-hCNGA3 (AGTC-402) กำลังดำเนินการ
ในการทดลอง NCT 03001310 (AAV8-hCARp.hCNGB3) ในผู้ใหญ่ 11 คนและเด็ก 12 คน รวม 23 คน ความปลอดภัยอยู่ในระดับที่ยอมรับได้ พบการมองเห็น สีดีขึ้นใน 6/23 คน อาการกลัวแสง ดีขึ้นใน 11/20 คน และคุณภาพชีวิตดีขึ้นใน 21/23 คน ในขนาดสูงพบแนวโน้มการเพิ่มขึ้นของการอักเสบภายในลูกตา 2)
ในการทดลอง AAV8.CNGA3 (RD-CURE) ผู้เข้าร่วม 9 คนได้รับสามขนาด (1×10¹⁰ ถึง 1×10¹¹ vg/ตา) ข้อมูลที่ 1 ปีและ 3 ปีแสดงแนวโน้มการปรับปรุงความคมชัดของภาพและความไวต่อคอนทราสต์ แต่ไม่มีนัยสำคัญทางสถิติ ความปลอดภัยดี 1) 2)
McKyton และคณะ (2021) ทำการทำแผนที่คอร์เทกซ์ด้วย fMRI หลังการฉีด AAV2tYF-PR1.7-hCNGA3 (NCT 02935517) ใต้จอประสาทตา ในผู้ใหญ่สองคนที่มี CNGA3-ACHM 4) ตาที่ได้รับการรักษาแสดงความทนทานต่อแสงมากกว่าตาที่ไม่ได้รับการรักษาถึง 5 เท่า (อาการกลัวแสง ดีขึ้นอย่างมาก) ได้รับความสามารถในการตรวจจับสีแดง และขนาด population receptive field (pRF) ลดลง บ่งชี้ถึงความละเอียดเชิงพื้นที่ที่ดีขึ้น ในทางกลับกัน ไม่พบการกระตุ้นบริเวณคอร์เทกซ์เฉพาะสี (เช่น V4) และการตรวจคลื่นไฟฟ้าจอประสาทตา แบบเต็มลานยังคงตรวจไม่พบการตอบสนองของเซลล์รูปกรวย ผู้ป่วยรายงานการปรับปรุงในชีวิตประจำวัน เช่น “ความรู้สึกปลอดภัยมากขึ้นเมื่อข้ามถนน” “ไม่จำเป็นต้องใช้แว่นขยาย” และ “ไม่จำเป็นต้องใช้แว่นกันแดดกลางแจ้ง”
การฟื้นฟูการทำงานหลังการเสริมยีนได้รับการยืนยันในแบบจำลองสัตว์หลายชนิด 1) 2)
แบบจำลองหนู : ฟื้นฟูได้ถึง 80% ของปกติในการตรวจคลื่นไฟฟ้าจอประสาทตา แบบจำลองสุนัข (CNGB3) : ฟื้นฟูการตรวจคลื่นไฟฟ้าจอประสาทตา แบบกระพริบของเซลล์รูปกรวย หลังติดตามผล 2.5 ปีหลังเสริมยีน พฤติกรรมดีขึ้นในสภาพแวดล้อม >25 ลักซ์แบบจำลองแกะ (CNGA3) : การปรับปรุงระยะยาวอย่างน้อย 6 ปีหลังเสริมยีนไพรเมตที่ไม่ใช่มนุษย์ (PDE6C) : ยืนยันการฟื้นฟูการทำงาน
เกี่ยวกับกรอบเวลาที่มีประสิทธิภาพของการรักษา การทดลองในสัตว์แสดงให้เห็นว่าการรักษาในวัยเด็กมีประสิทธิภาพมากกว่า หนูสูงอายุตอบสนองได้ไม่ดี แต่การเตรียมด้วย CNTF (ปัจจัยประสาทโทรฟิกซิลิอารี) ทำให้สามารถฟื้นฟูการทำงานในสุนัขสูงอายุได้ 1) 2)
การทดลองทางคลินิก (NCT 04041232) ของ phenylbutyrate glycerol (PBA) ที่กำหนดเป้าหมายการตอบสนองต่อความเครียดของเอนโดพลาสมิกเรติคูลัมกำลังดำเนินอยู่ 2) แนวทางนี้เป็นที่น่าสนใจในฐานะทางเลือกแทนการเสริมยีนโดยการบรรเทาการพับผิดในเอนโดพลาสมิกเรติคูลัม
ความท้าทายหลักของการบำบัดด้วยยีน มีดังนี้ 1) 2) 4)
ภูมิคุ้มกัน : การตอบสนองทางภูมิคุ้มกันต่อแคปซิด AAV อาจจำกัดประสิทธิภาพของการรักษาการปรับขนาดยาให้เหมาะสม : จำเป็นต้องสร้างสมดุลระหว่างประสิทธิภาพการรักษาและความเสี่ยงของการอักเสบภายในลูกตาความท้าทายด้านพัฒนาการ : ความยากในการผ่าตัดในดวงตาของเด็ก การจัดการกับการเกิดภาวะตาขี้เกียจ ข้อจำกัดของความยืดหยุ่นของคอร์เทกซ์การเห็น : ในผู้ใหญ่ การปรับโครงสร้างการทำงานของคอร์เทกซ์อาจทำได้ยาก ดังนั้นช่วงเวลาของการรักษาจึงสำคัญประสิทธิภาพระยะยาวและต้นทุน : การรับประกันผลที่ยั่งยืนและความท้าทายทางเศรษฐกิจด้านสุขภาพ
Q
การบำบัดด้วยยีนสามารถรักษาโรคตาบอดสีทั้งหมดให้หายขาดได้หรือไม่?
A
ปัจจุบันอยู่ในขั้นตอนการทดสอบความปลอดภัยและประสิทธิภาพระยะที่ I/II มีรายงานการลดอาการกลัวแสง และการปรับปรุงความไวแสงบางส่วน แต่ยังไม่สามารถฟื้นฟูการมองเห็น สีได้อย่างสมบูรณ์2) 4) การศึกษาในสัตว์ทดลองแสดงให้เห็นว่าการรักษาในวัยเด็กอาจมีประสิทธิภาพ แต่ข้อมูลระยะยาวในมนุษย์ยังมีจำกัด สิ่งสำคัญคือต้องติดตามความก้าวหน้าของการวิจัยและอัปเดตข้อมูลกับแพทย์ผู้รักษาอย่างสม่ำเสมอ
Michalakis S, Gerhardt M, Rudolph G, Priglinger S, Priglinger C. Achromatopsia: Genetics and Gene Therapy. Molecular diagnosis & therapy. 2022;26(1):51-59. doi:10.1007/s40291-021-00565-z. PMID:34860352; PMCI D:PMC8766373.
Baxter MF, Borchert GA. Gene Therapy for Achromatopsia. Int J Mol Sci. 2024;25(17):9739. doi:10.3390/ijms25179739.
Khan HM, Sumita FAG , Preti RC, Sarraf D, Navajas EV. CNGA3-Related Achromatopsia: A 10-Year Follow-Up. Journal of vitreoretinal diseases. 2026. doi:10.1177/24741264251414135. PMID:41626077; PMCI D:PMC12851907.
McKyton A, Averbukh E, Marks Ohana D, Levin N, Banin E. Cortical Visual Mapping following Ocular Gene Augmentation Therapy for Achromatopsia. J Neurosci. 2021;41(35):7363-7371. doi:10.1523/JNEUROSCI .3222-20.2021. PMID:34349002; PMCI D:PMC8412991.
Madeira C, Godinho G, Grangeia A, Falcão M, Silva R, Carneiro Â, et al. Two Novel Disease-Causing Variants in the PDE6C Gene Underlying Achromatopsia. Case reports in ophthalmology. 2021;12(3):749-760. doi:10.1159/000512284. PMID:34720973; PMCI D:PMC8460892.
Kohl S, Baumann B, Dassie F, et al. Paternal Uniparental Isodisomy of Chromosome 2 in a Patient with CNGA3-Associated Autosomal Recessive Achromatopsia. Int J Mol Sci. 2021;22(15):7842. doi:10.3390/ijms22157842. PMID:34360608; PMCI D:PMC8346044.
Rasmussen DK, Sun YJ, Franco JA, et al. Structure-function analysis of CNGA3-associated achromatopsia patient variants complements clinical genomics in pathogenicity determination. Orphanet J Rare Dis. 2025;20:261. doi:10.1186/s13023-025-03792-3.