CNGA3
کروموزوم: 2q11.2
عملکرد: زیرواحد آلفای کانال CNG
فراوانی: حدود 25 تا 50٪ موارد 1)2)
الگوی جهش: عمدتاً جهشهای نادرست (missense). دامنه گذرنده S4 یک نقطه داغ است.
پراکندگی جغرافیایی: در خاورمیانه و چین، CNGA3 بیش از 80٪ موارد را تشکیل میدهد.

آکروماتوپسی (Achromatopsia; ACHM) یک بیماری نادر ارثی شبکیه دوطرفه است که در آن عملکرد هر سه نوع سلول مخروطی از بین میرود. به آن «تکرنگی میلهای (rod monochromatism)» و «کوررنگی کامل (total color blindness)» نیز گفته میشود1).
شیوع این بیماری در سراسر جهان حدود یک در ۳۰,۰۰۰ نفر تخمین زده میشود1). الگوی توارث آن اتوزومال مغلوب است، با ناهنجاریهای سیستمیک همراه نیست و امید به زندگی طبیعی است.
ACHM دو نوع دارد: کامل (complete) و ناقص (incomplete). در نوع کامل، تمام عملکرد مخروطی از بین رفته است؛ در نوع ناقص، حداقل یک زیرگروه مخروطی عملکرد باقیمانده دارد و حدت بینایی حدود ۲۰/۴۰ تا ۲۰/۱۲۰ است و فوتوفوبی و نیستاگموس خفیفتر هستند2).
شش ژن عامل (CNGA3، CNGB3، GNAT2، PDE6C، PDE6H و ATF6) شناسایی شدهاند و در بیش از ۹۰٪ موارد میتوان ژن عامل را تعیین کرد1)2). تنها دو ژن CNGA3 و CNGB3 حدود ۸۰ تا ۹۰٪ موارد را تشکیل میدهند.
توجه داشته باشید که کوررنگی معمولی (نقص مادرزادی رنگبینی) ناشی از ناهنجاری در یک یا دو نوع ماده بینایی مخروطی است و فقط بر دید رنگ تأثیر میگذارد. ACHM از این جهت اساساً متفاوت است که همه مخروطها کار نمیکنند و با کاهش بینایی، نیستاگموس و فتوفوبیا همراه است.
یک مورد شناخته شده به عنوان اثر بنیانگذار در جزیره پینگلاپ (میکرونزی) وجود دارد. پس از طوفانی در دهه 1700 که جمعیت جزیره را به شدت کاهش داد، جهش CNGB3 (p.S435F) در میان ساکنان گسترش یافت و شیوع به حدود 10% و نرخ ناقل به حدود 30% رسید 1)2).
کوررنگی مادرزادی معمولی تنها تغییر در دید رنگ به دلیل ناهنجاری در یک یا دو نوع ماده بینایی مخروطی است و حدت بینایی در محدوده طبیعی است. آکروماتوپسی به دلیل فقدان عملکرد هر سه نوع مخروط، علاوه بر از دست دادن دید رنگ، با کاهش شدید بینایی (حدود 0.1 یا کمتر)، نیستاگموس، فتوفوبیا و همرالوپی همراه است که اساساً متفاوت است.
علائم ACHM در عرض چند هفته پس از تولد ظاهر میشوند.
واکنش متناقض مردمک (paradoxical pupil response): انقباض اولیه مردمک در تاریکی، یک یافته مشخصه. سرنخ مهمی برای تشخیص در کودکان است.
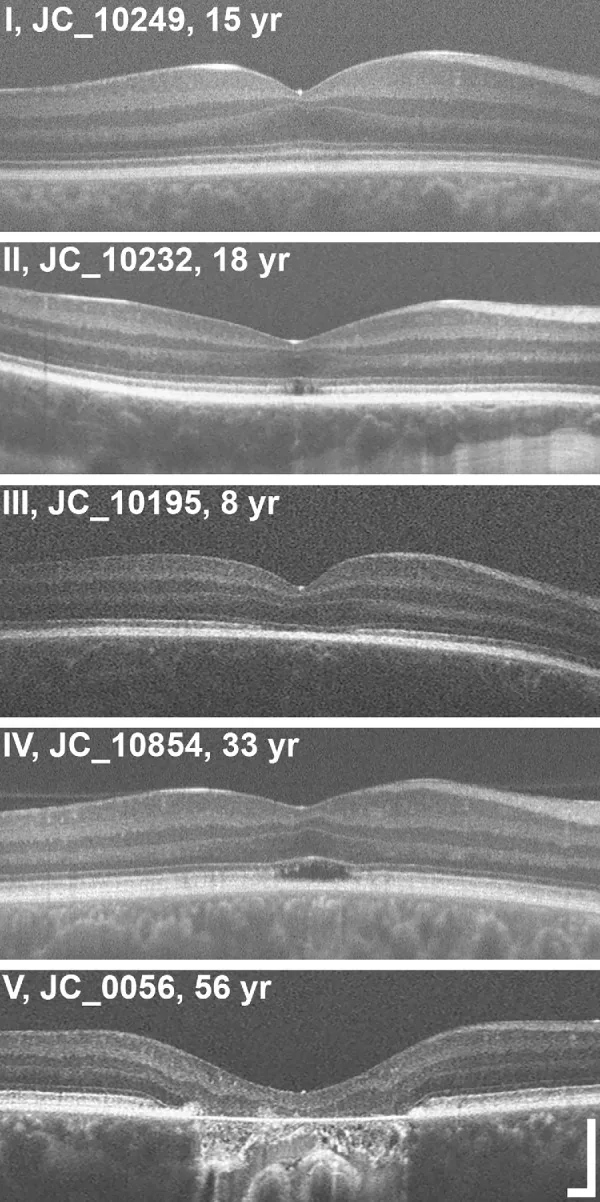
یافتههای فوندوس: در مراحل اولیه تقریباً طبیعی به نظر میرسد. آنژیوگرافی فلورسئین نیز تقریباً طبیعی است. با گذشت زمان، تغییرات لکهای و آتروفی در اپیتلیوم رنگدانه شبکیه (RPE) ممکن است رخ دهد. فقدان رفلکس ماکولا و دژنراسیون ماکولا اغلب دیده میشود. در OCT، اتصال داخلی-خارجی بخشهای گیرنده نوری (ellipsoid zone) و نوک بخش خارجی مخروطها (interdigitation zone) نامنظم و نامشخص است.
یافتههای الکترورتینوگرافی: در الکترورتینوگرافی روشنایی، پاسخ مخروطی به شدت کاهش یافته یا از بین رفته است، در حالی که در الکترورتینوگرافی تاریکی، پاسخ استوانهای طبیعی تا تقریباً طبیعی است1)2). در نوع GNAT2، مخروطهای S نسبت به نوع CNGA3/CNGB3 نسبتاً حفظ میشوند.
هیپوپلازی فووئال (foveal hypoplasia): در 60 تا 70 درصد موارد با جهشهای CNGA3/CNGB3 دیده میشود2).
FAF (خودفلورسانس فوندوس): چهار الگو وجود دارد: طبیعی، افزایش سیگنال مرکزی، کاهش سیگنال مرکزی، حلقه هیپرفلورسنت با هیپوفلورسانس مرکزی2).
مرحلهبندی OCT: تغییرات ساختاری لایههای خارجی فووئا در پنج مرحله ارزیابی میشود3).
| مرحله | یافته OCT |
|---|---|
| مرحله 1 | حفظ لایههای خارجی شبکیه (هایپررفلکتیویته ELM، مسطح شدن EZ) |
| مرحله 2 | تخریب نوار بیضوی (EZ) |
| مرحله 3 | ظهور فضای خالی نوری (optically empty space) |
| مرحله 4 | فضای خالی نوری + تخریب جزئی RPE |
| مرحله 5 | از بین رفتن لایه هسته خارجی و/یا تخریب کامل RPE |
در یک مطالعه پیگیری 10 ساله، با وجود ثبات بهترین حدت بینایی اصلاح شده (20/400 تا 20/200)، پیشرفت ساختاری در OCT (افزایش فضای خالی نوری: چشم راست 246×59 میکرومتر، چشم چپ 326×53 میکرومتر) تأیید شده است3).
نقاط پرنور (hyperreflective foci) بیش از 3 سال قبل از تغییرات EZ ظاهر میشوند و ممکن است نشانگر اولیه پیشرفت بیماری باشند3).
AOSLO (افتالموسکوپ لیزری اسکن تطبیقی): در موزاییک مخروطی، نقاط تاریک (dark space)، افزایش فاصله بین مخروطی و کاهش تراکم مخروطی مشاهده میشود. تفاوت معنیداری بین نوع CNGA3 و CNGB3 وجود ندارد و در نوع GNAT2 مخروطها نسبتاً حفظ شدهاند2).
تست رنگ: در صفحات رنگی ایشیهارا، به جز صفحه نمایش، تقریباً غیرقابل خواندن است. در پانل D-15، الگوی خطا در محور اسکوتوپیک (بین دوتان و تریتان) نشان میدهد. در آنومالوسکوپ، شیب تند نشان میدهد و محدوده تطابق رنگ طبیعی را شامل نمیشود.
بهترین حدت بینایی اصلاح شده اغلب در بلندمدت نسبتاً پایدار است. با این حال، ارزیابی ساختاری با OCT ممکن است تغییرات پیشرونده (تخریب لایه بیضوی، گسترش فضای خالی نوری) را نشان دهد3). جدایی بین عملکرد و ساختار مشخصه این بیماری است و پایش منظم با آزمایشهای دورهای اهمیت دارد.
وراثت اتوزومال مغلوب است. اگر هر دو والد ناقل باشند، خطر ابتلا برای فرزند 25٪ است1). معمولاً از والدین سالم ظاهری به ارث میرسد و اغلب سابقه خانوادگی وجود ندارد.
الگوی وراثت غیر مندلی ناشی از دیزومی تکوالد پدری (UPD) نیز گزارش شده است. در یک مورد که هموزیگوت برای CNGA3 c.778G>C (p.D260H) به دلیل UPD ایجاد شده بود، جهش در مادر یافت نشد 6). چنین مواردی برای ارزیابی خطر عود در مشاوره ژنتیک اهمیت دارند.
CNGA3
کروموزوم: 2q11.2
عملکرد: زیرواحد آلفای کانال CNG
فراوانی: حدود 25 تا 50٪ موارد 1)2)
الگوی جهش: عمدتاً جهشهای نادرست (missense). دامنه گذرنده S4 یک نقطه داغ است.
پراکندگی جغرافیایی: در خاورمیانه و چین، CNGA3 بیش از 80٪ موارد را تشکیل میدهد.
CNGB3
کروموزوم: 8q21.3
عملکرد: زیرواحد بتای کانال CNG
فراوانی: حدود 50٪ موارد 1)2)
الگوی جهش: عمدتاً جهشهای بیهوده (nonsense)، تغییر چارچوب (frameshift) و پیرایش (splicing). c.1148delC شایعترین جهش است.
پراکندگی جغرافیایی: در اروپا و آمریکا، CNGB3 بیش از 50٪ موارد را تشکیل میدهد.
سایر ژنها
GNAT2 (1p13.3): آلفای ترانسدوسین مخروطی. حدود 2%. نسبتاً خفیف با حفظ لایه گیرنده نوری 2)
PDE6C (10q23.33): زیرواحد آلفای PDE مخروطی. نوع شدید با شروع زودرس 5)
PDE6H (12p12.3): زیرواحد گامای PDE مخروطی. بسیار نادر 1)
ATF6 (1q23.3): فاکتور رونویسی پاسخ به استرس شبکه آندوپلاسمی. حدود 2%. مکانیسمی که مستقیماً در انتقال نور دخیل نیست 1)2)
آلل با نفوذ کم: CNGB3 c.1208G>A (p.R403Q) به دلیل حفظ عملکرد جزئی، منجر به فنوتیپ خفیف میشود 2).
ویژگیهای جهش PDE6C: در 4 مورد با جهشهای جدید (c.1670G>A، c.2192G>A)، سهگانه نیستاگموس، فوتوفوبی و اختلال دید رنگی در همه موارد مشاهده شد. الکترورتینوگرافی فقدان پاسخ فتوپیک و 30 هرتز فلیکر و پاسخ اسکوتوپیک طبیعی را تأیید کرد و هتروزیگوتهای مرکب فنوتیپ شدیدتری نشان دادند 5).
وراثت دیژنیک: موارد نادری با جهش در هر دو CNGA3 و CNGB3 وجود دارد 1).
از آنجا که آکروماتوپسی یک بیماری اتوزومال مغلوب است، حتی اگر هر یک از والدین یک نسخه از ژن جهشیافته را به عنوان ناقل داشته باشند، از نظر ظاهری سالم و دید رنگی طبیعی دارند. در ترکیب دو ناقل، با احتمال 25% فرزند دو نسخه جهش را دریافت کرده و مبتلا میشود 1).
در صورت مشاهده نیستاگموس، فوتوفوبی و کاهش بینایی در هفتههای اول پس از تولد، نیاز به بررسی جامع با در نظر گرفتن ACHM است. آزمایش ژنتیکی برای تشخیص قطعی ضروری است.
الکترورتینوگرافی
اهمیت تشخیصی: استاندارد طلایی
یافتههای نوع کامل: در الکترورتینوگرافی روشنایی، پاسخ مخروطی از بین رفته یا به شدت کاهش یافته است. در الکترورتینوگرافی تاریکی، پاسخ استوانهای طبیعی تا تقریباً طبیعی است1)2)
یافتههای نوع ناقص: پاسخ مخروطی ضعیف متناظر با عملکرد مخروطی باقیمانده تشخیص داده میشود
ویژگیهای نوع GNAT2: مخروطهای S نسبت به نوع CNGA3/CNGB3 نسبتاً حفظ شدهاند
OCT
آزمایش ژنتیکی
اهمیت تشخیصی: برای تشخیص قطعی ضروری است
روش: پانل هدفمند ۶ ژن (CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)2)
نرخ تشخیص: در بیش از ۹۰٪ موارد میتوان ژن عامل را شناسایی کرد1)
شرکت در کارآزمایی بالینی: تشخیص مولکولی برای شرکت در کارآزمایی بالینی ژن درمانی ضروری است
تشخیص افتراقی از بیماریهایی که تصویر بالینی مشابه ACHM دارند مهم است2).
در حال حاضر هیچ درمان قطعی تأیید شدهای برای ACHM وجود ندارد1)2). درمان عمدتاً علامتی است.
اگرچه دوربینی شایع است، اما طیف وسیعی از عیوب انکساری دیده میشود، بنابراین اصلاح با عینک یا لنز تماسی برای به حداکثر رساندن بینایی مهم است. در صورت وجود تنبلی چشم، درمان با پوشاندن چشم یا آتروپین در نظر گرفته شود.
لنزهای محافظ نور برای کاهش حساسیت به نور کیفیت زندگی روزمره را به طور قابل توجهی بهبود میبخشند. بر اساس یک مطالعه، 96٪ از بیماران فیلتر خاکستری را به فیلتر قرمز ترجیح میدهند و در فضای باز 74٪ فیلتر خاکستری را ترجیح میدهند2). انتخاب بر اساس ترجیحات فردی بیمار و محیط فعالیت مهم است.
ابزارها و فناوریهای زیر استفاده میشوند3).
توضیح و حمایت متخصص در مورد ویژگیهای وراثت اتوزومال مغلوب، خطر عود و اهمیت تشخیص ژنتیکی ضروری است. همچنین توصیه میشود برای شرکت در کارآزماییهای بالینی آینده ژن درمانی، تشخیص ژنتیکی انجام شود.
در یک نظرسنجی از بیماران، 96% فیلتر خاکستری را به فیلتر قرمز ترجیح دادند و در فضای باز 74% فیلتر خاکستری را ترجیح دادند2). با این حال، تفاوتهای فردی وجود دارد، بنابراین توصیه میشود فیلترهای مختلف را امتحان کرده و با مشورت چشمپزشک یا متخصص کمبینایی، مناسبترین را برای محیط فعالیت خود انتخاب کنید.
آبشار انتقال نور در سلولهای مخروطی طبیعی به شرح زیر است1).
جهش CNGA3: جهشهای missense باعث اختلال در تاخوردگی پروتئین، انتقال درون سلولی و ادغام در غشا میشوند1)7). دامنه گذرنده S4 یک نقطه داغ جهش است. بیش از 150 نوع جهش missense گزارش شده است که 103 مورد از نظر بیماریزایی تأیید نشده بودند، اما تحلیل ساختار سهبعدی-عملکرد نشان میدهد که 86.4% از آنها پیامدهای عملکردی مشابهی با جهشهای بیماریزای شناخته شده دارند7).
جهش CNGB3: جهشهای nonsense و frameshift منجر به تولید پروتئینهای کوتاه شده یا از دست رفته عملکرد کانال میشوند1). در غیاب CNGB3، کانالهای همومر CNGA3 باقی میمانند که ممکن است عملکرد مخروطی اندکی حفظ شود.
جهش ATF6: یک فاکتور رونویسی درگیر در پاسخ به پروتئینهای بدتاخورده شبکه آندوپلاسمی (UPR) است و مستقیماً در آبشار انتقال نور نقش ندارد1)2). مکانیسم پاتولوژیک آن با سایر ژنها متفاوت است و هنوز در حال بررسی است.
کانال CNG یک ساختار تترامری (CNGA3 × 3 و CNGB3 × 1، در برخی گزارشها 2:2) دارد و هر زیرواحد شامل شش دامنه گذرنده، یک دامنه اتصال نوکلئوتید حلقوی، ناحیه C-linker و دامنه تشکیلدهنده منفذ است1).
رشد و مورفولوژی اصلی مخروطها پس از تولد تقریباً طبیعی است و تصور میشود دژنراسیون از اوایل بزرگسالی شروع میشود. تجمع cGMP در فرآیند دژنراسیون نقش دارد و در مدلهای حیوانی، پیشرفت سریعتری در نواحی غنی از مخروط S نشان داده شده است1).
به طور سنتی، ACHM یک بیماری غیرپیشرونده در نظر گرفته میشد. با این حال، مشاهده طولانیمدت با OCT نشان داده است که با وجود تقریباً ثابت ماندن بهترین دید اصلاح شده، تغییرات ساختاری (تخریب EZ و گسترش فضای نوری) پیشرفت میکند3). این واگرایی عملکرد-ساختار در ارزیابی پنجره درمانی ژن درمانی اهمیت دارد.
در ناحیه بدون میله (rod-free zone) حفره مرکزی که ناحیه متراکم مخروطیها و فاقد میلههاست، از دست رفتن مخروطها قابل توجه است، در حالی که در ناحیه اطراف حفره مرکزی، میلهها با کمک به نوار EZ عملکرد را جبران میکنند 3).
کارآزماییهای بالینی اصلی ژن درمانی که تا سال 2024 در حال انجام یا تکمیل شدهاند در زیر نشان داده شدهاند 1)2).
| شماره NCT | ژن هدف | ناقل | وضعیت |
|---|---|---|---|
| NCT03001310 | CNGB3 | AAV8-hCARp.hCNGB3 | تکمیل شده |
| NCT02610582 | CNGA3 | AAV8-hG1.7-hCNGA3 | تکمیل شده |
| NCT03758404 | CNGA3 | rAAV8.hCNGA3 | در حال ثبت نام |
| NCT02599922 | CNGB3 | AAV2tYF-PR1.7-hCNGB3 (AGTC-401) | در حال انجام |
| NCT02935517 | CNGA3 | AAV2tYF-PR1.7-hCNGA3 (AGTC-402) | در حال انجام |
در کارآزمایی NCT03001310 (AAV8-hCARp.hCNGB3) روی 11 بزرگسال و 12 کودک (مجموعاً 23 نفر)، ایمنی در محدوده قابل قبول بود. بهبود دید رنگی در 6/23 نفر، بهبود حساسیت به نور در 11/20 نفر و بهبود کیفیت زندگی در 21/23 نفر مشاهده شد. در دوزهای بالا، تمایل به افزایش التهاب داخل چشمی نیز مشاهده شد 2).
در کارآزمایی AAV8.CNGA3 (RD-CURE)، 9 نفر سه دوز (1×10¹⁰ تا 1×10¹¹ vg/چشم) دریافت کردند. دادههای یک و سه ساله روند بهبود در حدت بینایی و حساسیت کنتراست را نشان داد، اما به significance آماری نرسید. ایمنی خوب بود 1)2).
McKyton و همکاران (2021) پس از تزریق زیرشبکیهای AAV2tYF-PR1.7-hCNGA3 (NCT02935517) در دو بزرگسال مبتلا به CNGA3-ACHM، نقشهبرداری قشر بینایی با fMRI انجام دادند 4). در چشم درمانشده نسبت به چشم درماننشده، تحمل نور 5 برابر (بهبود چشمگیر حساسیت به نور)، توانایی تشخیص رنگ قرمز و کاهش اندازه میدان دریافتی جمعیت (pRF) (نشاندهنده بهبود وضوح فضایی) تأیید شد. با این حال، فعالسازی مناطق خاص قشر بینایی برای رنگ (مانند V4) مشاهده نشد و در الکترورتینوگرافی تماممیدان نیز پاسخ مخروطی قابل تشخیص نبود. بیماران خودگزارشی بهبود در فعالیتهای روزمره مانند «احساس امنیت بیشتر در خط عابر پیاده»، «عدم نیاز به ذرهبین» و «عدم نیاز به عینک آفتابی در فضای باز» را گزارش کردند.
در چندین مدل حیوانی، بهبود عملکرد پس از جایگزینی ژن تأیید شده است 1)2).
در مورد پنجره زمانی مؤثر درمان، آزمایشهای حیوانی نشان داده است که درمان در سنین پایینتر مؤثرتر است. موشهای مسن پاسخ ضعیفی داشتند، اما پیشدرمان با CNTF (فاکتور نوروتروفیک مژگانی) امکان بهبود عملکرد را حتی در سگهای مسن فراهم کرد 1)2).
کارآزمایی بالینی فنیلبوتیرات گلیسرول (PBA) با هدف پاسخ به استرس شبکه آندوپلاسمی (NCT04041232) در حال انجام است 2). این رویکرد به جای جایگزینی ژن، بر کاهش نادرست تاشدگی در شبکه آندوپلاسمی متمرکز است.
چالشهای اصلی ژن درمانی به شرح زیر است 1)2)4).
در حال حاضر، این درمان در مرحله کارآزمایی فاز I/II ایمنی و اثربخشی است. کاهش حساسیت به نور و بهبود نسبی حساسیت نوری گزارش شده است، اما بازیابی کامل بینایی رنگ محقق نشده است2)4). مطالعات حیوانی نشان داده است که درمان در سنین پایین ممکن است مؤثرتر باشد، اما دادههای بلندمدت انسانی هنوز محدود است. پیگیری پیشرفت تحقیقات و بهروزرسانی منظم اطلاعات با پزشک معالج اهمیت دارد.