CNGA3
染色體: 2q11.2
功能: CNG通道α亞基
頻率: 約佔病例的25–50% 1)2)
突變模式: 以錯義突變為主。S4跨膜結構域是熱點區域。
地理分佈: 在中東和中國,CNGA3佔80%以上。

全色盲(ACHM)是一種罕見的雙側遺傳性視網膜疾病,所有三種錐狀感光細胞功能喪失。也稱為桿體單色視或完全色盲 1)。
全球盛行率估計約為每30,000人中有1人 1)。為體染色體隱性遺傳,不伴全身異常,預期壽命正常。
ACHM分為完全型和不完全型兩種。完全型所有錐狀功能缺失;不完全型至少一種錐狀亞型保留部分功能,視力約為20/40至20/120,畏光和眼球震顫較輕 2)。
已發現6種致病基因(CNGA3、CNGB3、GNAT2、PDE6C、PDE6H、ATF6),90%以上的病例可確定致病基因 1)2)。僅CNGA3和CNGB3就佔所有病例的80-90%。
需要注意的是,一般的色覺異常(先天性色覺異常)是由1~2種錐體視蛋白異常引起的,僅影響色覺。ACHM則因所有錐體細胞功能喪失,本質上不同,伴有視力下降、眼球震顫和畏光。
有一個著名的案例是平格拉普環礁(密克羅尼西亞)的創始者效應。在18世紀颱風導致人口銳減後,CNGB3突變(p.S435F)在島民中傳播,患病率約10%,攜帶率約30%1)2)。
ACHM的症狀在出生後數週內開始出現。
反常瞳孔反應: 在暗處出現瞳孔初始收縮的特徵性表現。是兒童診斷的重要線索。
眼底所見: 初期幾乎正常。螢光眼底造影也幾乎正常。隨著病程進展,可能出現視網膜色素上皮(RPE)的斑片狀變化和萎縮。常可見黃斑反射消失和黃斑變性。OCT顯示視網膜外層橢圓體帶和嵌合體帶不規則且模糊。
視網膜電圖所見: 明適應視網膜電圖顯示錐體反應顯著降低或消失,暗適應視網膜電圖顯示桿體反應正常或接近正常1)2)。GNAT2型中S錐體比CNGA3/CNGB3型相對保留。
中心凹發育不全: 在CNGA3/CNGB3突變中60-70%可見2)。
FAF(眼底自發螢光): 存在四種模式:正常、中心訊號增強、中心訊號減弱、高螢光環伴中心低螢光2)。
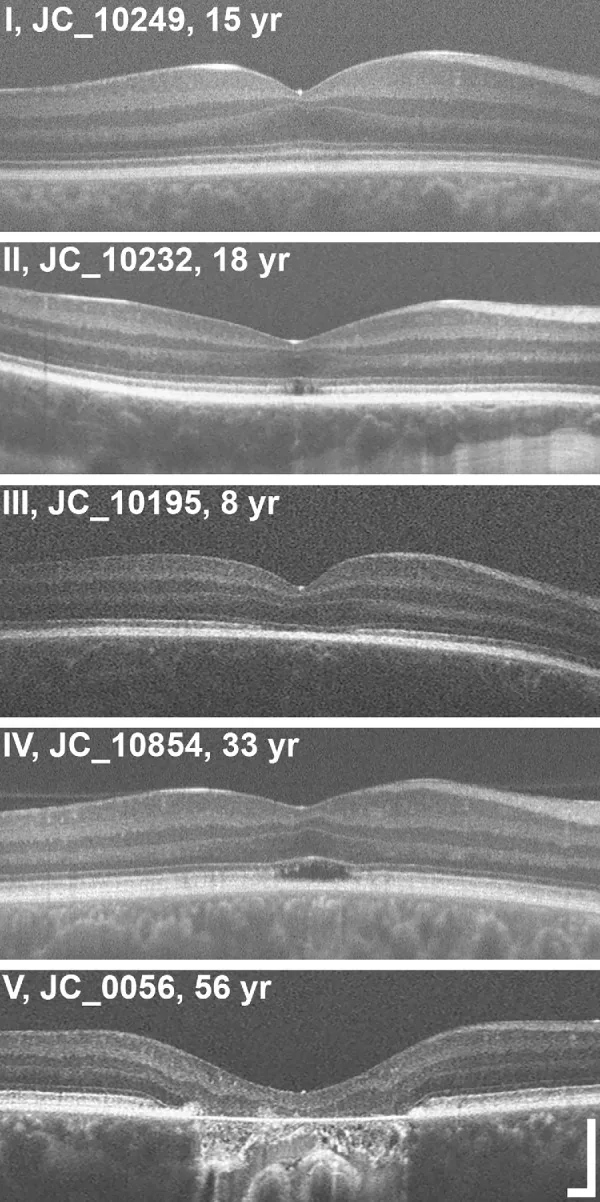
OCT分期: 中心凹外層結構變化分為5個階段進行評估3)。
| 分期 | OCT所見 |
|---|---|
| 第1期 | 外層視網膜保留(ELM高反射、EZ扁平化) |
| 第2期 | 橢圓體帶(EZ)破壞 |
| 第3期 | 出現光學空腔(optically empty space) |
| 第4期 | 光學空腔 + 部分RPE破壞 |
| 第5期 | 外核層消失和/或完全RPE破壞 |
在一項為期10年的追蹤研究中,儘管最佳矯正視力穩定(20/400至20/200),但OCT顯示結構性進展(光學空腔擴大:右眼246×59 μm,左眼326×53 μm)3)。
高反射灶在橢圓體帶變化前3年以上出現,提示其可能是疾病進展的早期標誌物3)。
AOSLO(自適應光學掃描雷射檢眼鏡):視錐細胞鑲嵌中出現暗區、視錐細胞間距增大、密度降低。CNGA3型和CNGB3型之間無顯著差異;GNAT2型視錐細胞相對保留2)。
色覺檢查:石原色盲檢查表除演示表外幾乎無法辨認。Panel D-15顯示暗視軸錯誤模式(介於紅色盲和藍色盲之間)。異常色覺鏡檢查顯示陡峭斜率,不包含正常匹配範圍。
最佳矯正視力通常長期穩定。然而,OCT的結構性評估可能顯示隨時間進展的變化(橢圓體帶破壞、光學空腔擴大)3)。功能與結構之間出現分離是其特點,定期監測很重要。
體染色體隱性遺傳。如果父母雙方均為攜帶者,子女發病風險為25%1)。患者通常由外表健康的父母所生,家族史可能缺失。
也有報告因父源單親二倍體(UPD)導致的非孟德爾遺傳模式。在一例因UPD導致CNGA3 c.778G>C (p.D260H)純合子的病例中,母親未檢測到突變6)。此類例子對遺傳諮詢中的復發風險評估具有重要意義。
CNGA3
染色體: 2q11.2
功能: CNG通道α亞基
頻率: 約佔病例的25–50% 1)2)
突變模式: 以錯義突變為主。S4跨膜結構域是熱點區域。
地理分佈: 在中東和中國,CNGA3佔80%以上。
CNGB3
染色體: 8q21.3
功能: CNG通道β亞基
頻率: 約佔病例的50% 1)2)
突變模式: 以無義、移碼和剪接突變為主。c.1148delC是最常見的突變。
地理分佈: 在歐洲和美國,CNGB3佔50%以上。
其他基因
GNAT2(1p13.3):錐體轉導素α亞基。約占2%。病情相對較輕,光感受器層保留2)
PDE6C(10q23.33):錐體PDE α亞基。早發型重症5)
PDE6H(12p12.3):錐體PDE γ亞基。極為罕見1)
ATF6(1q23.3):內質網壓力反應轉錄因子。約占2%。機制不直接參與光傳導1)2)
低外顯率等位基因:CNGB3 c.1208G>A (p.R403Q)保留部分功能,導致輕度表現型2)。
PDE6C突變特徵:在帶有新突變(c.1670G>A、c.2192G>A)的4例患者中,所有病例均出現眼球震顫、畏光和色覺障礙三聯徵。視網膜電圖顯示明視和30 Hz閃爍反應消失,暗視反應正常,複合雜合子表現出更嚴重的表現型5)。
雙基因遺傳:存在罕見的CNGA3和CNGB3均帶有突變的病例1)。
由於全色盲是體染色體隱性遺傳,父母可能各帶有一個突變基因拷貝(帶因者),外表健康且色覺正常。當兩個帶因者生育時,孩子有25%的機率繼承兩個突變拷貝而發病1)。
如果在出生後數週內出現眼球震顫、畏光和視力下降,需要進行以ACHM為重點的多方面檢查。確診必須依靠基因檢測。
視網膜電圖
OCT
基因檢測
診斷意義: 確診所必需
方法: 6個基因(CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)的目標面板2)
解決率: 超過90%的病例可確定致病基因1)
臨床試驗參與: 參加基因治療臨床試驗必須進行分子診斷
與具有類似臨床表現的疾病進行鑑別很重要2)。
目前,ACHM尚無批准的根治性治療方法1)2)。治療以對症治療為主。
遠視常見,但伴有廣泛的屈光不正。通過眼鏡或隱形眼鏡矯正對最大化視力很重要。如果合併弱視,可考慮遮蓋療法或阿托品治療。
用於減輕畏光的遮光鏡片可顯著改善生活品質。患者調查顯示,96%的人更喜歡灰色濾光片而不是紅色濾光片,74%的人在戶外更喜歡灰色濾光片2)。應根據患者個人偏好和活動環境進行選擇。
使用以下輔助器具與技術3)。
需要專家針對體染色體隱性遺傳的特性、復發風險以及基因診斷的意義進行說明與支援。也建議接受基因診斷,為將來參加基因治療臨床試驗做準備。
患者調查顯示,96%偏好灰色濾光片而非紅色濾光片,戶外74%偏好灰色濾光片2)。但由於個體差異,建議實際嘗試各種濾光片後,與眼科醫師或低視力專家協商選擇適合自己活動環境的濾光片。
正常錐狀感光細胞中的光傳導級聯如下1)。
CNGA3突變: 錯義突變損害蛋白質摺疊、細胞內運輸和膜整合1)7)。S4跨膜結構域是突變熱點。已報導超過150種錯義突變,其中103種致病性未確定,但三維結構-功能分析顯示86.4%與已知致病突變具有相似的功能後果7)。
CNGB3突變: 無義和框架位移突變產生截短或功能喪失的通道蛋白1)。CNGB3缺失時,殘留的CNGA3同源通道可能保留部分錐體功能。
ATF6突變: ATF6是參與內質網未摺疊蛋白反應(UPR)的轉錄因子,不直接參與光傳導級聯1)2)。其致病機轉與其他基因不同,目前仍在研究中。
CNG通道為四聚體結構(CNGA3 × 3和CNGB3 × 1,部分報告為2:2),每個亞單元包含6個跨膜結構域、環核苷酸結合結構域、C-linker區和孔道形成結構域1)。
出生後錐體的主要發育和形態接近正常,變性被認為始於青年期。cGMP累積參與變性過程,動物模型顯示S錐體豐富區域進展更快1)。
傳統上,ACHM被認為是一種非進展性疾病。然而,OCT長期觀察顯示,儘管最佳矯正視力大致穩定,但結構性變化(EZ破壞、光學空隙擴大)仍在進展3)。這種功能-結構分離對評估基因治療的治療窗口具有重要意義。
在中心凹的無桿體區(無桿體的錐體密集區域),錐體喪失顯著,而在旁中心凹,桿體透過貢獻於EZ帶而代償功能3)。
截至2024年正在進行或已完成的主要基因治療臨床試驗如下所示1)2)。
| NCT編號 | 標靶基因 | 載體 | 狀態 |
|---|---|---|---|
| NCT03001310 | CNGB3 | AAV8-hCARp.hCNGB3 | 已完成 |
| NCT02610582 | CNGA3 | AAV8-hG1.7-hCNGA3 | 已完成 |
| NCT03758404 | CNGA3 | rAAV8.hCNGA3 | 招募中 |
| NCT02599922 | CNGB3 | AAV2tYF-PR1.7-hCNGB3 (AGTC-401) | 進行中(尚未招募) |
| NCT02935517 | CNGA3 | AAV2tYF-PR1.7-hCNGA3 (AGTC-402) | 進行中(尚未招募) |
NCT03001310(AAV8-hCARp.hCNGB3)試驗共納入23名受試者(11名成人,12名兒童),安全性在可接受範圍內。6/23名受試者色覺改善,11/20名畏光改善,21/23名生活品質(QoL)改善。高劑量組觀察到眼內發炎增加的趨勢2)。
AAV8.CNGA3(RD-CURE)試驗中,9名受試者接受了三種劑量(1×10¹⁰至1×10¹¹ vg/眼)。1年和3年數據顯示視力和對比敏感度有改善趨勢,但未達到統計學顯著性。安全性良好1)2)。
McKyton等人(2021)對兩名CNGA3-ACHM成人患者進行AAV2tYF-PR1.7-hCNGA3(NCT02935517)視網膜下注射後,透過fMRI進行皮質視覺映射4)。治療眼與非治療眼相比,光耐受性提高5倍(畏光顯著改善),獲得紅色檢測能力,群體感受野(pRF)大小縮小(提示空間解析度提高)。然而,未觀察到顏色特異性皮質區域(如V4)的激活,全視野視網膜電圖仍檢測不到錐體反應。患者自我報告日常生活改善,如「過馬路安全感提高」、「無需放大鏡」、「戶外無需戴太陽眼鏡」。
多個動物模型已證實基因補充後的功能恢復1)2)。
關於治療的有效時間窗,動物實驗顯示年輕時期治療更有效。老年小鼠反應不佳,但CNTF(睫狀神經營養因子)前處理可使老年犬也能恢復功能1)2)。
針對內質網壓力反應的苯丁酸甘油酯(PBA)臨床試驗(NCT04041232)正在進行中2)。作為一種緩解內質網摺疊異常而非基因補充的方法,備受關注。
基因治療的主要挑戰如下1)2)4)。
目前處於第I/II期安全性與有效性試驗階段。已有報告顯示畏光減輕和部分光敏感性改善,但尚未實現完全色覺恢復2)4)。動物實驗表明,年輕時治療可能有效,但人類長期數據仍然有限。重要的是關注研究進展,並與主治醫師定期更新資訊。