Mù màu toàn bộ (Achromatopsia; ACHM) là một bệnh võng mạc di truyền hiếm gặp ở cả hai mắt, trong đó cả ba loại tế bào hình nón đều mất chức năng. Bệnh còn được gọi là “mù màu đơn sắc hình que” hoặc “mù màu hoàn toàn” 1).
Tỷ lệ mắc ước tính khoảng 1/30.000 người trên toàn thế giới 1). Bệnh di truyền theo kiểu lặn trên nhiễm sắc thể thường, không kèm bất thường toàn thân và tuổi thọ trung bình bình thường.
ACHM có hai thể: hoàn toàn và không hoàn toàn. Ở thể hoàn toàn, chức năng tế bào hình nón hoàn toàn không có; ở thể không hoàn toàn, ít nhất một loại tế bào hình nón còn chức năng tồn dư, thị lực khoảng 20/40 đến 20/120, và sợ ánh sáng cùng rung giật nhãn cầu nhẹ hơn 2).
Có sáu gen gây bệnh (CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, ATF6), và có thể xác định gen gây bệnh ở hơn 90% trường hợp 1)2). Riêng hai gen CNGA3 và CNGB3 chiếm 80-90% tổng số ca.
Cần lưu ý rằng các rối loạn thị giác màu sắc thông thường (rối loạn thị giác màu bẩm sinh) là do bất thường ở một hoặc hai sắc tố thị giác của tế bào hình nón và chỉ ảnh hưởng đến thị giác màu. ACHM khác biệt cơ bản vì tất cả các tế bào hình nón đều không hoạt động, kèm theo giảm thị lực, rung giật nhãn cầu và sợ ánh sáng.
Có một trường hợp nổi tiếng là hiệu ứng người sáng lập ở đảo Pingelap (Micronesia). Sau một cơn bão vào những năm 1700, dân số trên đảo giảm mạnh, và đột biến CNGB3 (p.S435F) lan rộng trong những người sống sót, đạt tỷ lệ mắc khoảng 10% và tỷ lệ người mang gen khoảng 30%1)2).
QSự khác biệt giữa mù màu toàn bộ và rối loạn thị giác màu thông thường (yếu màu) là gì?
A
Các rối loạn thị giác màu bẩm sinh thông thường là do bất thường ở một hoặc hai sắc tố thị giác của tế bào hình nón và chỉ ảnh hưởng đến thị giác màu, với thị lực bình thường. Trong mù màu toàn bộ, chức năng của cả ba tế bào hình nón đều thiếu hụt, dẫn đến mất thị giác màu, kèm theo giảm thị lực (khoảng 0,1 hoặc thấp hơn), rung giật nhãn cầu, sợ ánh sáng và quáng gà, khác biệt cơ bản.
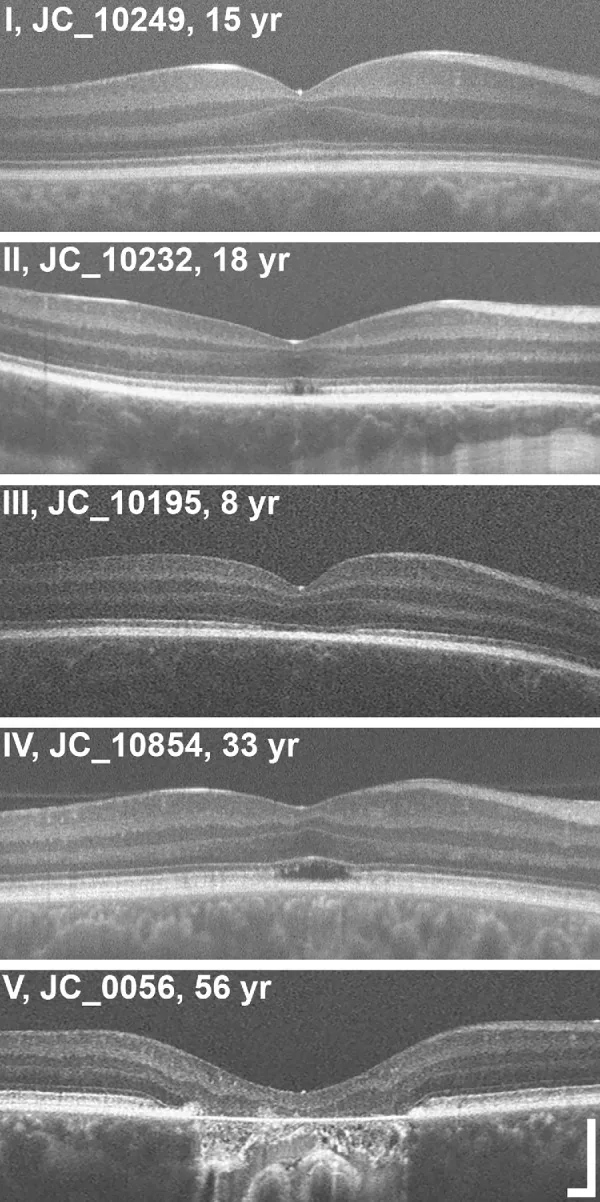
Grissim G, et al. Longitudinal Assessment of OCT-Based Measures of Foveal Cone Structure in Achromatopsia. Invest Ophthalmol Vis Sci. 2024. Figure 1. PMCID: PMC11005076. License: CC BY.
Phân loại năm mức độ EZ bằng OCT, cho thấy EZ bình thường (I), gián đoạn EZ (II), biến mất EZ (III), vùng phản xạ thấp (IV) và teo võng mạc ngoài cùng RPE (V). Tương ứng với các bất thường vùng ellipsoid được thảo luận trong phần “2. Các triệu chứng chính và dấu hiệu lâm sàng”.
Các triệu chứng của ACHM bắt đầu xuất hiện trong vài tuần đầu sau sinh.
Giảm thị lực: Ở thể hoàn toàn, dưới 20/200 (khoảng 0,1); ở thể không hoàn toàn, khoảng 20/80 (khoảng 0,25).
Sợ ánh sáng (nhạy cảm với ánh sáng): Trong một khảo sát bệnh nhân, 38% cho biết đây là triệu chứng nghiêm trọng nhất2). Chức năng thị giác giảm đáng kể trong môi trường sáng.
Quáng gà (hemeralopia): Chức năng thị giác giảm ở nơi sáng, nhưng tương đối tốt ở môi trường tối.
Mất/giảm thị lực màu: Mất thị lực màu ở cả ba trục1). Trong bảng kiểm tra Ishihara, hầu như không đọc được ngoại trừ bảng trình diễn. Xét nghiệm Panel D-15 cho thấy các đường nhầm lẫn hình tròn xiên.
Rung giật nhãn cầu hình con lắc: Xuất hiện trong vài tuần đầu sau sinh. Có xu hướng cải thiện khi lớn lên và giảm khi nhìn gần.
Kết hợp với viễn thị: Thường gặp viễn thị, nhưng có thể kèm theo nhiều tật khúc xạ khác bao gồm cận thị.
Phản ứng đồng tử nghịch thường: Co đồng tử ban đầu trong bóng tối. Dấu hiệu đặc trưng quan trọng trong chẩn đoán trẻ em.
Kết quả đáy mắt: Ban đầu gần như bình thường. Chụp mạch huỳnh quang cũng cho thấy kết quả gần như bình thường. Theo thời gian, có thể xuất hiện các thay đổi dạng đốm và teo biểu mô sắc tố võng mạc. Thường thấy mất phản xạ hoàng điểm hoặc thoái hóa hoàng điểm. Trên OCT, chỗ nối giữa đoạn trong và đoạn ngoài của tế bào cảm thụ và vùng xen kẽ không đều và không rõ.
Kết quả điện võng mạc: Trên ERG sáng, đáp ứng của tế bào nón giảm rõ rệt hoặc mất, trong khi đáp ứng của tế bào que trên ERG tối bình thường hoặc gần bình thường1)2). Ở thể GNAT2, tế bào nón S được bảo tồn tương đối tốt hơn so với thể CNGA3/CNGB3.
FAF (Huỳnh quang tự phát đáy mắt): Có bốn dạng: bình thường, tăng tín hiệu trung tâm, giảm tín hiệu trung tâm, vòng tăng huỳnh quang kèm giảm huỳnh quang trung tâm2).
Phân giai đoạn OCT: Đánh giá các thay đổi cấu trúc của lớp ngoài hố trung tâm theo năm giai đoạn3).
Giai đoạn
Kết quả OCT
Giai đoạn 1
Bảo tồn các lớp ngoài võng mạc (tăng phản xạ ELM, làm phẳng EZ)
Giai đoạn 2
Phá hủy vùng ellipsoid (EZ)
Giai đoạn 3
Xuất hiện khoang quang học rỗng (optically empty space)
Giai đoạn 4
Khoang quang học rỗng + phá hủy một phần RPE
Giai đoạn 5
Mất lớp nhân ngoài và/hoặc phá hủy hoàn toàn RPE
Trong một nghiên cứu theo dõi 10 năm, mặc dù thị lực tốt nhất đã chỉnh kính ổn định (20/400 đến 20/200), nhưng tiến triển cấu trúc trên OCT (mở rộng khoang quang học rỗng: mắt phải 246×59 μm, mắt trái 326×53 μm) đã được xác nhận3).
Các điểm tăng phản xạ (hyperreflective foci) có thể xuất hiện hơn 3 năm trước những thay đổi của EZ, cho thấy tiềm năng là dấu hiệu sớm của tiến triển bệnh3).
AOSLO (Kính soi đáy mắt quét laser quang học thích ứng): Khảm tế bào hình nón cho thấy vùng tối (dark space), khoảng cách giữa các tế bào hình nón tăng lên và mật độ tế bào hình nón giảm. Không có sự khác biệt đáng kể giữa loại CNGA3 và CNGB3, trong khi loại GNAT2 bảo tồn tế bào hình nón tương đối tốt2).
Kiểm tra thị lực màu sắc: Trên bảng Ishihara, hầu như tất cả các bảng đều không đọc được ngoại trừ bảng trình diễn. Trên Panel D-15, cho thấy mô hình lỗi trên trục scotopic (giữa deutan và tritan). Trên anomaloscope, cho thấy độ dốc cao và không bao gồm phạm vi cân bằng màu bình thường.
QThị lực của bệnh mù màu toàn bộ có xấu đi theo tuổi tác không?
A
Thị lực tốt nhất đã chỉnh kính thường ổn định trong thời gian dài. Mặt khác, đánh giá cấu trúc bằng OCT có thể cho thấy những thay đổi theo thời gian (phá hủy vùng ellipsoid, mở rộng khoang quang học rỗng) có thể tiến triển3). Có sự phân ly giữa chức năng và cấu trúc, và việc theo dõi thường xuyên bằng các xét nghiệm định kỳ là rất quan trọng.
Di truyền lặn trên nhiễm sắc thể thường. Nếu cả bố và mẹ đều là người mang gen (carrier), nguy cơ con mắc bệnh là 25%1). Bệnh thường xảy ra ở trẻ em có cha mẹ bề ngoài khỏe mạnh và trong nhiều trường hợp không có tiền sử gia đình.
Một kiểu di truyền không Mendel do thể đơn bội hai nguồn (UPD) từ cha cũng đã được báo cáo. Trong trường hợp đồng hợp tử về CNGA3 c.778G>C (p.D260H) được hình thành qua UPD, không phát hiện đột biến ở mẹ 6). Những ví dụ như vậy có ý nghĩa quan trọng trong đánh giá nguy cơ tái phát ở tư vấn di truyền.
Kiểu đột biến: Đột biến sai nghĩa chiếm ưu thế. Vùng xuyên màng S4 là điểm nóng
Phân bố địa lý: Ở Trung Đông và Trung Quốc, CNGA3 chiếm hơn 80%
CNGB3
Nhiễm sắc thể: 8q21.3
Chức năng: Tiểu đơn vị beta của kênh CNG
Tần suất: Khoảng 50% các trường hợp1)2)
Kiểu đột biến: Đột biến vô nghĩa, dịch khung và nối ghép chiếm ưu thế. c.1148delC là đột biến phổ biến nhất
Phân bố địa lý: Ở châu Âu và châu Mỹ, CNGB3 chiếm hơn 50%
Các gen khác
GNAT2 (1p13.3): Transducin α của tế bào nón. Khoảng 2%. Tương đối nhẹ, lớp tế bào cảm thụ ánh sáng được bảo tồn 2)
PDE6C (10q23.33): Tiểu đơn vị α của PDE tế bào nón. Thể nặng khởi phát sớm 5)
PDE6H (12p12.3): Tiểu đơn vị γ của PDE tế bào nón. Rất hiếm 1)
ATF6 (1q23.3): Yếu tố phiên mã đáp ứng stress lưới nội chất. Khoảng 2%. Cơ chế không liên quan trực tiếp đến dẫn truyền ánh sáng 1)2)
Alen thâm nhập thấp: CNGB3 c.1208G>A (p.R403Q) giữ lại chức năng một phần, dẫn đến kiểu hình nhẹ 2).
Đặc điểm đột biến PDE6C: Ở 4 ca có đột biến mới (c.1670G>A, c.2192G>A), bộ ba (rung giật nhãn cầu, sợ ánh sáng, rối loạn thị giác màu) được ghi nhận ở tất cả. Điện võng mạc cho thấy mất thị lực ban ngày và nhấp nháy 30 Hz với thị lực ban đêm bình thường, và dị hợp tử kép cho thấy kiểu hình nặng hơn 5).
Di truyền lưỡng gen: Có những trường hợp hiếm mang đột biến ở cả CNGA3 và CNGB3 1).
QTại sao trẻ có thể bị mù màu toàn bộ mặc dù cả bố và mẹ đều không mắc bệnh?
A
Vì mù màu toàn bộ là bệnh lặn trên nhiễm sắc thể thường, cả bố và mẹ có thể mỗi người mang một bản sao gen đột biến (người mang) mà không có triệu chứng và thị giác màu bình thường. Khi hai người mang gen có con, xác suất đứa trẻ thừa hưởng hai bản sao đột biến và mắc bệnh là 25% 1).
Nếu quan sát thấy rung giật nhãn cầu, sợ ánh sáng và giảm thị lực trong vài tuần đầu sau sinh, cần tiến hành kiểm tra toàn diện với nghi ngờ ACHM. Xét nghiệm di truyền là cần thiết để chẩn đoán xác định.
Điện võng mạc
Ý nghĩa chẩn đoán: Tiêu chuẩn vàng
Kết quả của thể hoàn toàn: Phản ứng của tế bào nón trên điện võng mạc dưới ánh sáng sáng biến mất hoặc giảm nghiêm trọng. Phản ứng của tế bào que trên điện võng mạc dưới ánh sáng tối bình thường đến gần như bình thường1)2)
Kết quả của thể không hoàn toàn: Phát hiện phản ứng nón yếu tương ứng với chức năng nón còn lại
Đặc điểm của thể GNAT2: Tế bào nón S được bảo tồn tương đối tốt hơn so với thể CNGA3/CNGB3
OCT
Ý nghĩa chẩn đoán: Tiêu chuẩn để đánh giá cấu trúc
Nội dung đánh giá: Phân giai đoạn 5 mức độ của lớp ngoài hố mắt, phát hiện thiểu sản hố mắt2)3)
Theo dõi: Ngay cả khi chức năng ổn định, vẫn có thể có tiến triển về cấu trúc, do đó việc theo dõi định kỳ rất quan trọng
Xét nghiệm bổ trợ: Đánh giá đa phương thức kết hợp FAF và AOSLO
Xét nghiệm di truyền
Ý nghĩa chẩn đoán: Cần thiết để chẩn đoán xác định
Phương pháp: Bảng gen mục tiêu cho 6 gen (CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)2)
Tỷ lệ giải quyết: Có thể xác định gen gây bệnh trong hơn 90% trường hợp1)
Tham gia thử nghiệm lâm sàng: Chẩn đoán phân tử là bắt buộc để tham gia thử nghiệm lâm sàng liệu pháp gen
Mặc dù viễn thị thường gặp, nhưng có nhiều loại tật khúc xạ, do đó việc chỉnh kính hoặc kính áp tròng rất quan trọng để tối đa hóa thị lực. Nếu có nhược thị, hãy xem xét liệu pháp che mắt hoặc atropine.
Thấu kính chống sáng để giảm chứng sợ ánh sáng cải thiện đáng kể chất lượng cuộc sống hàng ngày. Dữ liệu khảo sát bệnh nhân cho thấy 96% ưa thích bộ lọc màu xám hơn màu đỏ, và 74% ưa thích bộ lọc màu xám khi ở ngoài trời2). Việc lựa chọn phù hợp với sở thích cá nhân và môi trường hoạt động của bệnh nhân là rất quan trọng.
Cần có sự giải thích và hỗ trợ từ chuyên gia về đặc điểm di truyền lặn nhiễm sắc thể thường, nguy cơ tái phát và ý nghĩa của chẩn đoán di truyền. Cũng nên thực hiện chẩn đoán di truyền để chuẩn bị cho các thử nghiệm lâm sàng liệu pháp gen trong tương lai.
QMàu sắc nào phù hợp cho kính chắn sáng?
A
Trong một khảo sát bệnh nhân, 96% ưa thích bộ lọc màu xám hơn bộ lọc màu đỏ, và 74% ưa thích bộ lọc màu xám khi ở ngoài trời 2). Tuy nhiên, do sự khác biệt cá nhân, nên thử nhiều loại bộ lọc khác nhau và chọn loại phù hợp với môi trường hoạt động của bạn sau khi tham khảo ý kiến bác sĩ nhãn khoa hoặc chuyên gia thị lực kém.
Dòng thác truyền tín hiệu ánh sáng trong tế bào hình nón bình thường như sau1).
Trong tối: Nồng độ cGMP nội bào cao, kênh CNG mở, Na⁺ và Ca²⁺ đi vào, tế bào duy trì trạng thái khử cực và giải phóng glutamate liên tục.
Khi tiếp xúc với ánh sáng: Opsin (sắc tố thị giác) được kích hoạt → Transducin (protein G) được kích hoạt → Phosphodiesterase (PDE) được kích hoạt → cGMP bị phân hủy → Kênh CNG đóng → Siêu phân cực → Ức chế giải phóng glutamate.
Phản hồi âm tính: GCAP (Protein hoạt hóa Guanylate Cyclase) liên kết với Ca²⁺ và ức chế hoạt động của retGC, điều chỉnh sản xuất cGMP.
Đột biến CNGA3: Đột biến sai nghĩa làm suy yếu quá trình gấp protein, vận chuyển nội bào và tích hợp màng1)7). Vùng xuyên màng S4 là điểm nóng đột biến. Hơn 150 đột biến sai nghĩa đã được báo cáo, 103 đột biến chưa xác định được tính gây bệnh, nhưng phân tích cấu trúc-chức năng 3D cho thấy 86,4% có hậu quả chức năng tương tự như các đột biến gây bệnh đã biết7).
Đột biến CNGB3: Đột biến vô nghĩa và dịch khung tạo ra protein kênh bị cắt ngắn hoặc mất chức năng1). Khi thiếu CNGB3, các kênh đồng nhất CNGA3 vẫn tồn tại, do đó có thể duy trì một số chức năng hình nón tối thiểu.
Đột biến ATF6: Là yếu tố phiên mã tham gia vào phản ứng protein sai gấp (UPR) của lưới nội chất, và không tham gia trực tiếp vào dòng thác truyền tín hiệu ánh sáng1)2). Cơ chế bệnh lý khác với các gen khác và vẫn đang được nghiên cứu.
Kênh CNG có cấu trúc tetramer (CNGA3 × 3 và CNGB3 × 1, trong một số báo cáo là 2:2), mỗi tiểu đơn vị có 6 vùng xuyên màng, vùng liên kết nucleotide vòng, vùng liên kết C và vùng tạo lỗ1).
Sự phát triển chính sau sinh của các tế bào hình nón và hình thái của chúng gần như bình thường, và sự thoái hóa được cho là bắt đầu từ đầu tuổi trưởng thành. Sự tích tụ cGMP có liên quan đến quá trình thoái hóa, và sự tiến triển nhanh hơn ở các vùng giàu tế bào hình nón S đã được chứng minh trên mô hình động vật1).
Trước đây, ACHM được coi là bệnh không tiến triển. Tuy nhiên, quan sát dài hạn bằng OCT đã chứng minh rằng mặc dù thị lực tốt nhất có điều chỉnh gần như ổn định, nhưng các thay đổi cấu trúc (phá hủy EZ, mở rộng khoảng trống quang học) vẫn tiến triển3). Sự phân ly chức năng-cấu trúc này có ý nghĩa quan trọng trong việc đánh giá cửa sổ điều trị cho liệu pháp gen.
Ở vùng không có tế bào que (vùng tập trung nhiều tế bào nón) của hố mắt, sự mất tế bào nón rõ rệt, trong khi ở vùng cận hố mắt, tế bào que bù trừ chức năng bằng cách đóng góp vào dải EZ3).
7. Nghiên cứu mới nhất và Triển vọng tương lai (Báo cáo giai đoạn nghiên cứu)
Trong thử nghiệm NCT03001310 (AAV8-hCARp.hCNGB3) trên 11 người lớn và 12 trẻ em (tổng 23 người), tính an toàn ở mức chấp nhận được. Cải thiện thị lực màu sắc ở 6/23, cải thiện chứng sợ ánh sáng ở 11/20, và cải thiện chất lượng cuộc sống ở 21/23. Ở liều cao, có xu hướng tăng viêm nội nhãn2).
Trong thử nghiệm AAV8.CNGA3 (RD-CURE), 9 người được dùng ba liều (1×10¹⁰ đến 1×10¹¹ vg/mắt). Dữ liệu 1 năm và 3 năm cho thấy xu hướng cải thiện thị lực và độ nhạy tương phản, nhưng không đạt ý nghĩa thống kê. Tính an toàn tốt 1)2).
McKyton và cộng sự (2021) đã thực hiện lập bản đồ vỏ não bằng fMRI sau khi tiêm dưới võng mạc AAV2tYF-PR1.7-hCNGA3 (NCT02935517) ở hai người lớn mắc CNGA3-ACHM 4). Mắt được điều trị cho thấy khả năng chịu ánh sáng cao gấp 5 lần so với mắt không được điều trị (cải thiện đáng kể chứng sợ ánh sáng), đạt được khả năng phát hiện màu đỏ, và giảm kích thước trường tiếp nhận quần thể (pRF) cho thấy cải thiện độ phân giải không gian. Mặt khác, không quan sát thấy sự kích hoạt các vùng vỏ não đặc hiệu màu sắc (như V4), và điện võng mạc toàn trường vẫn không phát hiện được phản ứng của tế bào hình nón. Bệnh nhân báo cáo cải thiện trong sinh hoạt hàng ngày như “cảm giác an toàn hơn khi băng qua đường”, “không cần kính lúp”, và “không cần kính râm khi ra ngoài”.
Sự phục hồi chức năng sau khi bổ sung gen đã được xác nhận trên nhiều mô hình động vật 1)2).
Mô hình chuột: Phục hồi tới 80% mức bình thường trên điện võng mạc.
Mô hình chó (CNGB3): Phục hồi điện võng mạc nhấp nháy tế bào hình nón sau 2,5 năm theo dõi sau bổ sung gen. Cải thiện hành vi trong môi trường >25 lux.
Mô hình cừu (CNGA3): Cải thiện lâu dài ít nhất 6 năm sau bổ sung gen.
Động vật linh trưởng không phải người (PDE6C): Xác nhận phục hồi chức năng.
Về cửa sổ thời gian hiệu quả của điều trị, các thí nghiệm trên động vật cho thấy điều trị ở độ tuổi trẻ hiệu quả hơn. Chuột già phản ứng kém, nhưng tiền xử lý với CNTF (yếu tố dinh dưỡng thần kinh thể mi) cho phép phục hồi chức năng ở chó già 1)2).
Phương pháp tiếp cận không dùng liệu pháp gen cho đột biến ATF6
Một thử nghiệm lâm sàng (NCT04041232) với phenylbutyrate glycerol (PBA) nhắm vào phản ứng stress lưới nội chất đang được tiến hành 2). Phương pháp này được chú ý như một giải pháp thay thế cho bổ sung gen bằng cách giảm bớt sự gấp cuộn sai trong lưới nội chất.
Những thách thức chính của liệu pháp gen như sau 1)2)4).
Tính sinh miễn dịch: Phản ứng miễn dịch đối với capsid AAV có thể hạn chế hiệu quả điều trị.
Tối ưu hóa liều lượng: Cần cân bằng giữa hiệu quả điều trị và nguy cơ viêm nội nhãn.
Thách thức phát triển: Khó khăn trong phẫu thuật mắt trẻ em, xử lý sự xuất hiện của nhược thị.
Giới hạn tính dẻo của vỏ não thị giác: Ở người lớn, việc tái tổ chức chức năng vỏ não có thể khó khăn, do đó thời điểm điều trị rất quan trọng.
Hiệu quả lâu dài và chi phí: Đảm bảo hiệu quả bền vững và các thách thức kinh tế y tế.
QLiệu liệu pháp gen có thể chữa khỏi hoàn toàn bệnh mù màu toàn bộ không?
A
Hiện tại, đang ở giai đoạn thử nghiệm an toàn và hiệu quả pha I/II. Đã có báo cáo về giảm sợ ánh sáng và cải thiện một phần độ nhạy sáng, nhưng chưa đạt được phục hồi hoàn toàn thị lực màu sắc2)4). Nghiên cứu trên động vật cho thấy điều trị ở độ tuổi trẻ có thể hiệu quả, nhưng dữ liệu dài hạn trên người vẫn còn hạn chế. Điều quan trọng là theo dõi tiến trình nghiên cứu và cập nhật thông tin thường xuyên với bác sĩ điều trị.
McKyton A, Averbukh E, Marks Ohana D, Levin N, Banin E. Cortical Visual Mapping following Ocular Gene Augmentation Therapy for Achromatopsia. J Neurosci. 2021;41(35):7363-7371. doi:10.1523/JNEUROSCI.3222-20.2021. PMID:34349002; PMCID:PMC8412991.
Madeira C, Godinho G, Grangeia A, Falcão M, Silva R, Carneiro Â, et al. Two Novel Disease-Causing Variants in the PDE6C Gene Underlying Achromatopsia. Case reports in ophthalmology. 2021;12(3):749-760. doi:10.1159/000512284. PMID:34720973; PMCID:PMC8460892.
Kohl S, Baumann B, Dassie F, et al. Paternal Uniparental Isodisomy of Chromosome 2 in a Patient with CNGA3-Associated Autosomal Recessive Achromatopsia. Int J Mol Sci. 2021;22(15):7842. doi:10.3390/ijms22157842. PMID:34360608; PMCID:PMC8346044.
Rasmussen DK, Sun YJ, Franco JA, et al. Structure-function analysis of CNGA3-associated achromatopsia patient variants complements clinical genomics in pathogenicity determination. Orphanet J Rare Dis. 2025;20:261. doi:10.1186/s13023-025-03792-3.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.