Akromatopsi (Achromatopsia; ACHM), üç tip koni fotoreseptör hücresinin işlevinin kaybolduğu nadir, iki taraflı kalıtsal bir retina hastalığıdır. Ayrıca «çubuk monokromatizmi (rod monochromatism)» ve «tam renk körlüğü (total color blindness)» olarak da adlandırılır1).
Dünya genelinde prevalansı yaklaşık 30.000’de 1 olarak tahmin edilmektedir1). Otozomal resesif kalıtım gösterir, sistemik anormallikler eşlik etmez ve yaşam süresi normaldir.
ACHM’nin tam (complete) ve eksik (incomplete) olmak üzere iki tipi vardır. Tam tipte tüm koni işlevi yoktur; eksik tipte en az bir koni alt tipi kısmi işleve sahiptir, görme keskinliği 20/40 ila 20/120 civarındadır ve fotofobi ile nistagmus daha hafiftir2).
Altı sorumlu gen (CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, ATF6) tanımlanmıştır ve vakaların %90’ından fazlasında genetik tanı mümkündür1)2). Sadece CNGA3 ve CNGB3 genleri tüm vakaların %80-90’ını oluşturur.
Normal renk körlüğünün (doğuştan renk görme bozukluğu) bir veya iki tür koni görme pigmentindeki anormallikten kaynaklandığını ve yalnızca renk görmeyi etkilediğini unutmayın. ACHM, tüm konilerin işlev görmemesi nedeniyle görme azalması, nistagmus ve fotofobi ile birlikte olması açısından temelde farklıdır.
Pingelap Adası (Mikronezya) kurucu etkisi olarak bilinen bir vaka vardır. 1700’lerdeki bir tayfunun ardından ada nüfusu ciddi şekilde azalmış ve CNGB3 mutasyonu (p.S435F) halk arasında yayılmış, prevalans yaklaşık %10’a, taşıyıcılık oranı ise yaklaşık %30’a ulaşmıştır 1)2).
QAkromatopsi ile normal renk körlüğü (renk zayıflığı) arasındaki fark nedir?
A
Normal doğuştan renk körlüğü, bir veya iki tür koni görme pigmentindeki anormallik nedeniyle yalnızca renk görmede değişikliktir ve görme keskinliği normal aralıktadır. Akromatopsi, her üç koni türünün de işlevinin olmaması nedeniyle renk görme kaybına ek olarak görme keskinliğinde azalma (yaklaşık 0.1 veya daha düşük), nistagmus, fotofobi ve hemeralopi ile birlikte olması açısından temelde farklıdır.
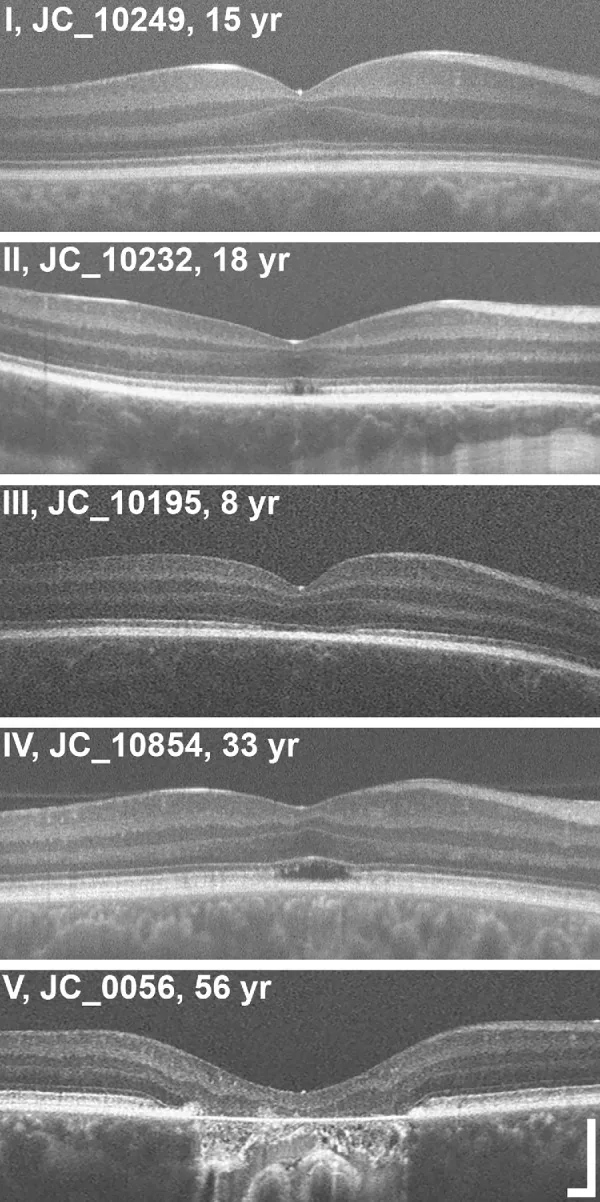
Grissim G, et al. Longitudinal Assessment of OCT-Based Measures of Foveal Cone Structure in Achromatopsia. Invest Ophthalmol Vis Sci. 2024. Figure 1. PMCID: PMC11005076. License: CC BY.
OCT ile EZ’nin beş aşamalı derece sınıflandırması: normal EZ (I), EZ kesintisi (II), EZ kaybı (III), düşük yansıma alanı (IV), dış retina atrofisi ve RPE (V). Bu, metnin “2. Başlıca belirtiler ve klinik bulgular” bölümünde ele alınan elipsoid bölge anormalliklerine karşılık gelir.
ACHM belirtileri doğumdan sonraki birkaç hafta içinde ortaya çıkmaya başlar.
Görme azalması: Tam tipte 20/200 (yaklaşık 0.1) veya daha düşük; eksik tipte yaklaşık 20/80 (yaklaşık 0.25).
Fotofobi (ışık hassasiyeti): Hasta anketinde %38’i en önemli belirti olarak bildirmiştir 2). Parlak ortamlarda görme işlevi belirgin şekilde azalır.
Hemeralopi (gündüz körlüğü): Aydınlık ortamda görme işlevi azalır, loş ortamlarda nispeten iyi görme keskinliği gösterir.
Renk görme yokluğu/azlığı: Üç eksende de renk görme kaybı1). İshihara renk görme testinde gösteri tablosu dışında neredeyse okunamaz. Panel D-15 testi dairesel çapraz karıştırma çizgileri gösterir.
Pendüler nistagmus: Doğumdan sonraki birkaç hafta içinde ortaya çıkar. Büyümeyle birlikte düzelme eğilimi gösterir ve yakın bakışta azalır.
Hipermetropi birlikteliği: Çoğunlukla hipermetropi görülür, ancak bazılarında miyopi dahil geniş bir kırma kusuru yelpazesi eşlik eder.
Paradoksal pupil yanıtı: Karanlıkta pupillanın başlangıçta daralması karakteristik bir bulgudur. Çocuklarda tanıda önemli bir ipucudur.
Fundus bulguları: Erken dönemde neredeyse normal görünür. Floresein anjiyografide de neredeyse normal bulgular gösterir. Zamanla retina pigment epitelinde (RPE) benekli değişiklikler ve atrofi gelişebilir. Makula refleksinin kaybı ve makula dejenerasyonu sıklıkla görülür. OCT’de retina dış tabakalarındaki fotoreseptör iç segment-dış segment bağlantısı (ellipsoid zone) ve koni dış segment ucu (interdigitation zone) düzensiz ve belirsizdir.
Elektroretinografi bulguları: Aydınlık ortam elektroretinografisinde koni yanıtı belirgin şekilde azalmış veya kaybolmuştur, karanlık ortam elektroretinografisinde ise çubuk yanıtı normal veya normale yakındır1)2). GNAT2 tipinde S konileri CNGA3/CNGB3 tipine göre nispeten korunmuştur.
FAF (Fundus otofloresansı): Dört patern mevcuttur: normal, merkezi sinyal artışı, merkezi sinyal azalması, hiperfloresan halka + merkezi hipofloresans2).
OCT evrelemesi: Fovea dış tabakalarındaki yapısal değişiklikler beş aşamada değerlendirilir3).
Evre
OCT bulgusu
Evre 1
Dış retina tabakalarının korunması (ELM hiperreflektivitesi, EZ düzleşmesi)
Evre 2
Elipsoid bandın (EZ) yıkımı
Evre 3
Optik boşluk (optically empty space) görünümü
Evre 4
Optik boşluk + kısmi RPE yıkımı
Evre 5
Dış nükleer tabaka kaybı ve/veya tam RPE yıkımı
10 yıllık bir takip çalışmasında, en iyi düzeltilmiş görme keskinliği stabil (20/400 ila 20/200) olmasına rağmen, OCT’de yapısal ilerleme (optik boşluk genişlemesi: sağ göz 246×59 μm, sol göz 326×53 μm) doğrulanmıştır3).
Hiperreflektif odaklar (hyperreflective foci), EZ değişikliklerinden 3 yıldan daha önce ortaya çıkar ve hastalık ilerlemesinin erken bir belirteci olabileceği öne sürülmüştür3).
AOSLO (Adaptif Optik Taramalı Lazer Oftalmoskop): Kon mozaiğinde karanlık alanlar (dark space), kon aralığında artış ve kon yoğunluğunda azalma görülür. CNGA3 ve CNGB3 tipleri arasında anlamlı fark yoktur, GNAT2 tipinde ise konlar nispeten korunmuştur2).
Renk testi: İshihara renk testi tablolarında, gösteri tablosu dışında neredeyse okunamaz. Panel D-15’te skotopik eksende (deutan ve tritan arasında) hata paterni gösterir. Anomaloskopta dik bir eğim gösterir ve normal renk eşleme aralığını içermez.
QAkromatopside görme yaşla birlikte kötüleşir mi?
A
En iyi düzeltilmiş görme keskinliği genellikle uzun vadede oldukça stabildir. Bununla birlikte, OCT ile yapısal değerlendirme yıllar içinde ilerleyici değişiklikler (ellipsoid bant yıkımı, optik boşluğun genişlemesi) gösterebilir3). Fonksiyon ve yapı arasında ayrışma karakteristiktir ve düzenli testlerle izlem önemlidir.
Otozomal resesif kalıtımdır. Her iki ebeveyn de taşıyıcı ise, çocuğun hastalığa yakalanma riski %25’tir1). Genellikle dışarıdan sağlıklı görünen ebeveynlerden doğar ve çoğu zaman aile öyküsü yoktur.
Paternal uniparental disomy (UPD) kaynaklı Mendel dışı kalıtım paterni de bildirilmiştir. CNGA3 c.778G>C (p.D260H) homozigotluğunun UPD ile oluştuğu bir vakada annede mutasyon saptanmamıştır 6). Bu tür örnekler, genetik danışmanlıkta tekrarlama riski değerlendirmesi açısından önemlidir.
Mutasyon paterni: Çoğunlukla missense mutasyonlar. S4 transmembran domaini sıcak noktadır.
Coğrafi dağılım: Orta Doğu ve Çin’de CNGA3 %80’in üzerindedir.
CNGB3
Kromozom: 8q21.3
İşlev: CNG kanalı beta alt birimi
Sıklık: Vakaların yaklaşık %50’si 1)2)
Mutasyon paterni: Çoğunlukla nonsense, frameshift ve splicing mutasyonları. c.1148delC en sık görülen mutasyondur.
Coğrafi dağılım: Avrupa ve Amerika’da CNGB3 %50’nin üzerindedir.
Diğer Genler
GNAT2 (1p13.3): Koni transdüsin alfa. Yaklaşık %2. Nispeten hafif seyirli, fotoreseptör tabakası korunmuş 2)
PDE6C (10q23.33): Koni PDE alfa alt birimi. Erken başlangıçlı şiddetli tip 5)
PDE6H (12p12.3): Koni PDE gama alt birimi. Son derece nadir 1)
ATF6 (1q23.3): Endoplazmik retikulum stres yanıtı transkripsiyon faktörü. Yaklaşık %2. Işık iletimine doğrudan katılmayan mekanizma 1)2)
Düşük penetranslı alel: CNGB3 c.1208G>A (p.R403Q) kısmi işlevi koruduğu için hafif fenotipe yol açar 2).
PDE6C mutasyonlarının özellikleri: Yeni mutasyonlara (c.1670G>A, c.2192G>A) sahip 4 olguda, nistagmus, fotofobi ve renk görme bozukluğu üçlüsü tüm olgularda gözlendi. Elektroretinografide fotopik ve 30 Hz flicker yanıtlarının kaybı ve skotopik yanıtların normal olduğu doğrulandı; bileşik heterozigotlar daha şiddetli fenotip gösterdi 5).
Digenik kalıtım: Hem CNGA3 hem de CNGB3’te mutasyon taşıyan nadir vakalar mevcuttur 1).
QEbeveynler akromatopsi değilken çocuğun neden hasta olduğu?
A
Akromatopsi otozomal resesif kalıtıldığı için, ebeveynlerin her biri bir kopya mutant gen taşıyıcısı olsa bile dışarıdan sağlıklı ve renk görüşü normaldir. Taşıyıcı çiftlerde %25 olasılıkla çocuk iki kopya mutasyonu alarak hastalanır 1).
Doğumdan sonraki ilk haftalarda nistagmus, fotofobi ve görme azlığı görüldüğünde ACHM düşünülerek kapsamlı inceleme gerekir. Kesin tanı için genetik test zorunludur.
Elektroretinografi
Tanısal önemi: Altın standart
Tam tip bulguları: Aydınlık ortam elektroretinografisinde koni yanıtı kaybolmuş veya belirgin şekilde azalmıştır. Karanlık ortam elektroretinografisinde çubuk yanıtı normal veya normale yakındır1)2)
Eksik tip bulguları: Kalan koni fonksiyonuna karşılık gelen zayıf bir koni yanıtı tespit edilir
GNAT2 tipinin özellikleri: S konileri CNGA3/CNGB3 tipine göre nispeten korunmuştur
Hipermetropi sık olmakla birlikte geniş bir refraksiyon kusuru yelpazesi görülür, bu nedenle gözlük veya kontakt lens ile düzeltme görme keskinliğini en üst düzeye çıkarmak için önemlidir. Ambliyopi eşlik ediyorsa kapama tedavisi veya atropin tedavisi düşünülmelidir.
Fotofobiyi azaltmak için ışık filtreli lensler günlük yaşam kalitesini önemli ölçüde iyileştirir. Bir hasta anketinde %96’sı gri filtreyi kırmızı filtreye tercih etmiş ve dış mekanda %74’ü gri filtreyi tercih etmiştir2). Bireysel hasta tercihlerine ve aktivite ortamına göre seçim yapmak önemlidir.
Otozomal resesif kalıtımın özellikleri, tekrarlama riski ve genetik tanının önemi hakkında uzman tarafından açıklama ve destek gereklidir. Gelecekteki gen tedavisi klinik çalışmalarına katılmak için genetik tanı yaptırılması da önerilir.
QIşık koruyucu gözlükler için hangi renk uygundur?
A
Hasta anketinde %96’sı gri filtreyi kırmızı filtreye tercih etmiş ve dış mekanda %74’ü gri filtreyi tercih etmiştir2). Ancak bireysel farklılıklar olduğu için, çeşitli filtreleri deneyerek kendi aktivite ortamınıza uygun olanı göz doktoru veya az görme uzmanı ile danışarak seçmeniz önerilir.
CNGA3 mutasyonu: Missense mutasyonlar protein katlanmasını, hücre içi taşınmasını ve membran entegrasyonunu bozar1)7). S4 transmembran domaini mutasyon sıcak noktasıdır. 150’den fazla missense mutasyon rapor edilmiş olup, 103 mutasyonun patojenitesi belirsizdi, ancak 3 boyutlu yapı-fonksiyon analizi, %86.4’ünün bilinen patojenik mutasyonlarla benzer fonksiyonel sonuçlara sahip olduğunu göstermiştir7).
CNGB3 mutasyonu: Nonsense ve çerçeve kayması mutasyonları, kesilmiş veya işlev kaybına uğramış kanal proteinleri üretir1). CNGB3 yokluğunda CNGA3 homomer kanalları kalır, bu nedenle az da olsa koni fonksiyonu korunabilir.
ATF6 mutasyonu: Endoplazmik retikulumda yanlış katlanmış protein yanıtında (UPR) rol oynayan bir transkripsiyon faktörüdür ve ışık iletim kaskadında doğrudan yer almaz1)2). Patolojik mekanizması diğer genlerden farklıdır ve halen araştırılmaktadır.
CNG kanalı tetramerik bir yapıya sahiptir (CNGA3 × 3 ve CNGB3 × 1, bazı raporlarda 2:2) ve her alt birim altı transmembran domain, bir siklik nükleotid bağlama domaini, C-linker bölgesi ve por oluşturan domain içerir1).
Koni dejenerasyonunun mekanizması ve ilerleyiciliği
Doğum sonrası konilerin ana gelişimi ve morfolojisi normale yakındır ve dejenerasyonun genç erişkinlik döneminde başladığı düşünülmektedir. cGMP birikiminin dejenerasyon sürecinde rol oynadığı ve hayvan modellerinde S-koni zengin bölgelerde daha hızlı ilerleme gösterildiği belirtilmiştir1).
Geleneksel olarak ACHM ilerleyici olmayan bir hastalık olarak kabul edilirdi. Ancak OCT ile uzun süreli gözlem, en iyi düzeltilmiş görme keskinliği neredeyse sabit kalmasına rağmen yapısal değişikliklerin (EZ bozulması, optik boşluğun genişlemesi) ilerlediğini göstermiştir3). Bu fonksiyon-yapı ayrışması, gen tedavisinin tedavi penceresini değerlendirmede önemlidir.
Foveadaki rodsuz bölgede (çubuk hücrelerin bulunmadığı koni yoğun alan) koni kaybı belirgindir, buna karşılık parafoveal bölgede çubuk hücreler EZ bandına katkıda bulunarak işlevi telafi eder 3).
7. Güncel Araştırmalar ve Gelecek Perspektifler (Araştırma Aşamasındaki Raporlar)
NCT03001310 (AAV8-hCARp.hCNGB3) çalışmasında 11 yetişkin ve 12 çocuk (toplam 23 kişi) üzerinde güvenlik kabul edilebilir sınırlar içindeydi. Renk görme iyileşmesi 6/23 kişide, fotofobi iyileşmesi 11/20 kişide ve yaşam kalitesi (QoL) iyileşmesi 21/23 kişide gözlendi. Yüksek dozlarda göz içi inflamasyonunda artış eğilimi de gözlendi 2).
AAV8.CNGA3 (RD-CURE) çalışmasında 9 kişiye üç doz (1×10¹⁰ ila 1×10¹¹ vg/göz) uygulandı. 1 yıl ve 3 yıl verilerinde görme keskinliği ve kontrast duyarlılığında iyileşme eğilimi görüldü ancak istatistiksel anlamlılığa ulaşmadı. Güvenlik iyiydi 1)2).
McKyton ve ark. (2021), CNGA3-ACHM’li iki yetişkine AAV2tYF-PR1.7-hCNGA3 (NCT02935517) subretinal enjeksiyonu sonrası fMRI ile kortikal görsel haritalama yaptı 4). Tedavi edilen gözde, tedavi edilmeyen göze kıyasla 5 kat ışık toleransı (fotofobide dramatik iyileşme), kırmızı algılama yeteneği kazanımı ve popülasyon reseptif alan (pRF) boyutunda küçülme (uzaysal çözünürlükte iyileşmeyi düşündüren) doğrulandı. Bununla birlikte, renge özgü kortikal alanlarda (V4 vb.) aktivasyon gözlenmedi ve tam alan elektroretinogramda koni yanıtı saptanamaz durumda kaldı. Hastaların kendi bildirimlerinde “yaya geçidinde güvenlik hissinde artış”, “büyüteç gereksiniminin kalmaması” ve “dışarıda güneş gözlüğüne ihtiyaç duymama” gibi günlük yaşam iyileşmeleri rapor edildi.
Birden fazla hayvan modelinde gen yerine koyma sonrası fonksiyonel iyileşme doğrulanmıştır 1)2).
Fare modeli: Elektroretinogramda normalin %80’ine kadar iyileşme.
Köpek modeli (CNGB3): Gen yerine koyma sonrası 2,5 yıllık takipte koni flaş elektroretinogram iyileşmesi. 25 lüks üzeri ortamlarda davranışsal iyileşme.
Koyun modeli (CNGA3): Gen yerine koyma sonrası en az 6 yıl süren uzun süreli iyileşme.
İnsan olmayan primat (PDE6C): Fonksiyonel iyileşme doğrulandı.
Tedavinin etkili zaman penceresi ile ilgili olarak, genç yaşta tedavinin daha etkili olduğu hayvan deneylerinde gösterilmiştir. Yaşlı farelerde yanıt zayıftı, ancak CNTF (siliyer nörotrofik faktör) ön tedavisi ile yaşlı köpeklerde bile fonksiyonel iyileşme mümkün oldu 1)2).
Endoplazmik retikulum stres yanıtını hedef alan fenilbütirat gliserol (PBA) klinik çalışması (NCT04041232) devam etmektedir 2). Gen yerine koyma yerine endoplazmik retikulumdaki katlanma bozukluğunu hafifletmeye yönelik bir yaklaşım olarak dikkat çekmektedir.
Gen tedavisinin başlıca zorlukları aşağıdaki gibidir 1)2)4).
İmmünojenite: AAV kapsidine karşı bağışıklık yanıtı tedavi etkinliğini sınırlayabilir.
Doz optimizasyonu: Tedavi etkinliği ile göz içi inflamasyon riski arasında denge sağlanması gerekir.
Gelişimsel zorluklar: Çocuk gözünde cerrahinin zorluğu ve ambliyopi gelişiminin yönetimi.
Görsel korteks plastisite sınırlamaları: Yetişkinlerde kortikal fonksiyonel yeniden yapılanma zor olabilir, tedavi zamanlaması önemlidir.
Uzun dönem etkinlik ve maliyet: Kalıcı etkinin garantisi ve sağlık ekonomisi zorlukları.
QGen tedavisi tam renk körlüğünü tedavi edebilir mi?
A
Şu anda, bu tedavi Faz I/II güvenlik ve etkinlik çalışması aşamasındadır. Fotofobide azalma ve kısmi ışık hassasiyeti iyileşmesi rapor edilmiştir, ancak tam renk görme geri kazanımı sağlanamamıştır2)4). Hayvan çalışmaları, genç yaşta tedavinin daha etkili olabileceğini göstermiştir, ancak insanlarda uzun dönem veriler hala sınırlıdır. Araştırmanın ilerlemesini takip etmek ve düzenli olarak sorumlu doktorla bilgi güncellemesi yapmak önemlidir.
McKyton A, Averbukh E, Marks Ohana D, Levin N, Banin E. Cortical Visual Mapping following Ocular Gene Augmentation Therapy for Achromatopsia. J Neurosci. 2021;41(35):7363-7371. doi:10.1523/JNEUROSCI.3222-20.2021. PMID:34349002; PMCID:PMC8412991.
Madeira C, Godinho G, Grangeia A, Falcão M, Silva R, Carneiro Â, et al. Two Novel Disease-Causing Variants in the PDE6C Gene Underlying Achromatopsia. Case reports in ophthalmology. 2021;12(3):749-760. doi:10.1159/000512284. PMID:34720973; PMCID:PMC8460892.
Kohl S, Baumann B, Dassie F, et al. Paternal Uniparental Isodisomy of Chromosome 2 in a Patient with CNGA3-Associated Autosomal Recessive Achromatopsia. Int J Mol Sci. 2021;22(15):7842. doi:10.3390/ijms22157842. PMID:34360608; PMCID:PMC8346044.
Rasmussen DK, Sun YJ, Franco JA, et al. Structure-function analysis of CNGA3-associated achromatopsia patient variants complements clinical genomics in pathogenicity determination. Orphanet J Rare Dis. 2025;20:261. doi:10.1186/s13023-025-03792-3.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.