Buta warna total (Akromatopsia; ACHM) adalah penyakit retina herediter bilateral langka di mana ketiga jenis sel kerucut kehilangan fungsinya. Juga disebut “monokromatisme batang” atau “buta warna total” 1).
Prevalensi diperkirakan sekitar 1 dari 30.000 orang di seluruh dunia 1). Penyakit ini diturunkan secara resesif autosomal, tidak disertai kelainan sistemik, dan harapan hidup normal.
ACHM terdiri dari dua tipe: komplit dan inkomplit. Pada tipe komplit, semua fungsi kerucut tidak ada; pada tipe inkomplit, setidaknya satu subtipe kerucut masih memiliki fungsi sisa, dengan ketajaman penglihatan sekitar 20/40 hingga 20/120, serta fotofobia dan nistagmus yang lebih ringan 2).
Gen penyebab ada enam (CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, ATF6), dan gen penyebab dapat diidentifikasi pada lebih dari 90% kasus 1)2). Dua gen, CNGA3 dan CNGB3, mencakup 80-90% dari seluruh kasus.
Perlu dicatat bahwa kelainan penglihatan warna umum (kelainan penglihatan warna bawaan) disebabkan oleh kelainan pada satu atau dua pigmen visual kerucut dan hanya memengaruhi penglihatan warna. ACHM berbeda secara fundamental karena semua kerucut tidak berfungsi, disertai penurunan ketajaman penglihatan, nistagmus, dan fotofobia.
Ada kasus terkenal sebagai efek pendiri di Pulau Pingelap (Mikronesia). Setelah topan pada tahun 1700-an, populasi pulau menurun drastis, dan mutasi CNGB3 (p.S435F) menyebar di antara penduduk yang selamat, mencapai prevalensi sekitar 10% dan tingkat karier sekitar 30%1)2).
QApa perbedaan antara buta warna total dan kelainan penglihatan warna umum (buta warna parsial)?
A
Kelainan penglihatan warna bawaan umum disebabkan oleh kelainan pada satu atau dua pigmen visual kerucut dan hanya memengaruhi penglihatan warna, dengan ketajaman penglihatan normal. Pada buta warna total, fungsi ketiga kerucut tidak ada, sehingga selain kehilangan penglihatan warna, juga disertai penurunan ketajaman penglihatan (sekitar 0,1 atau kurang), nistagmus, fotofobia, dan rabun senja, yang secara fundamental berbeda.
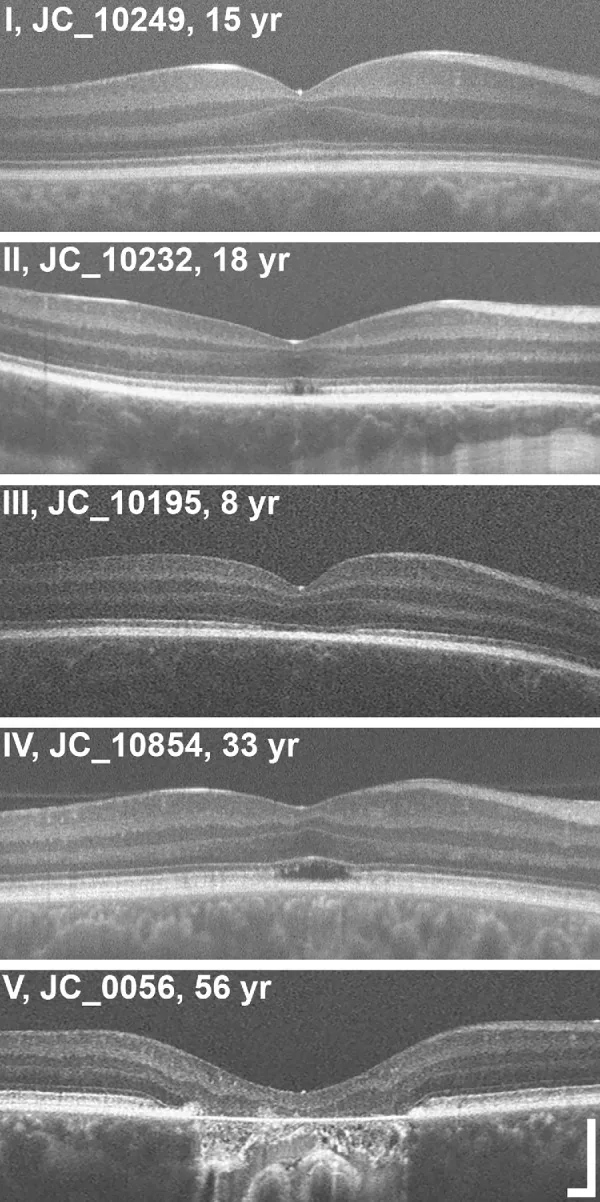
Grissim G, et al. Longitudinal Assessment of OCT-Based Measures of Foveal Cone Structure in Achromatopsia. Invest Ophthalmol Vis Sci. 2024. Figure 1. PMCID: PMC11005076. License: CC BY.
Klasifikasi lima grade EZ dengan OCT, menunjukkan EZ normal (I), diskontinuitas EZ (II), hilangnya EZ (III), area reflektifitas rendah (IV), dan atrofi retina luar serta RPE (V). Ini sesuai dengan kelainan zona ellipsoid yang dibahas di bagian “2. Gejala utama dan temuan klinis”.
Gejala ACHM mulai muncul dalam beberapa minggu pertama setelah lahir.
Penurunan ketajaman penglihatan: Pada tipe komplit, di bawah 20/200 (sekitar 0,1); pada tipe inkomplit, sekitar 20/80 (sekitar 0,25).
Fotofobia (sensitivitas cahaya): Dalam survei pasien, 38% melaporkannya sebagai gejala paling signifikan2). Fungsi penglihatan menurun drastis di lingkungan terang.
Rabun senja (hemeralopia): Fungsi penglihatan menurun di tempat terang, tetapi relatif baik di lingkungan redup.
Kehilangan/penurunan penglihatan warna: Hilangnya penglihatan warna pada ketiga sumbu1). Pada tes Ishihara, hampir semua pelat tidak terbaca kecuali pelat demonstrasi. Tes Panel D-15 menunjukkan garis kebingungan melingkar miring.
Nistagmus pendular: Muncul dalam beberapa minggu pertama setelah lahir. Cenderung membaik seiring pertumbuhan dan berkurang saat melihat dekat.
Asosiasi hipermetropia: Sering ditemukan hipermetropia, namun dapat disertai berbagai kelainan refraksi termasuk miopia.
Refleks pupil paradoks: Konstriksi pupil awal di tempat gelap. Temuan khas yang penting dalam diagnosis anak.
Temuan fundus: Awalnya tampak hampir normal. Angiografi fluorescein juga menunjukkan temuan hampir normal. Seiring waktu, dapat terjadi perubahan bercak dan atrofi epitel pigmen retina. Sering ditemukan tidak adanya refleks fovea atau degenerasi makula. Pada OCT, sambungan segmen dalam-luar fotoreseptor dan zona interdigitasi tidak teratur dan tidak jelas.
Temuan elektroretinogram: Pada ERG fotopik, respons kerucut menurun drastis atau menghilang, sedangkan respons batang pada ERG skotopik normal atau hampir normal1)2). Pada tipe GNAT2, kerucut S lebih terpelihara dibandingkan tipe CNGA3/CNGB3.
Hipoplasia foveal: Ditemukan pada 60-70% kasus mutasi CNGA3/CNGB32).
FAF (Autofluoresensi Fundus): Terdapat empat pola: normal, peningkatan sinyal sentral, penurunan sinyal sentral, cincin hiperfluoresen dengan hipofluoresen sentral2).
Staging OCT: Evaluasi perubahan struktural lapisan luar fovea dalam lima tahap3).
Stadium
Temuan OCT
Stadium 1
Pemeliharaan lapisan luar retina (hiperreflektif ELM, perataan EZ)
Stadium 2
Penghancuran zona ellipsoid (EZ)
Tahap 3
Munculnya ruang optik kosong (optically empty space)
Tahap 4
Ruang optik kosong + kerusakan RPE parsial
Tahap 5
Hilangnya lapisan inti luar dan/atau kerusakan RPE total
Dalam studi tindak lanjut 10 tahun, meskipun ketajaman visual terbaik terkoreksi stabil (20/400 hingga 20/200), perkembangan struktural pada OCT (perluasan ruang optik kosong: mata kanan 246×59 μm, mata kiri 326×53 μm) telah dikonfirmasi3).
Hiperreflektif fokus (hyperreflective foci) mungkin muncul lebih dari 3 tahun sebelum perubahan EZ, menunjukkan potensi sebagai penanda awal perkembangan penyakit3).
AOSLO (Adaptive Optics Scanning Laser Ophthalmoscope): Mosaik kerucut menunjukkan ruang gelap (dark space), peningkatan jarak antar kerucut, dan penurunan kepadatan kerucut. Tidak ada perbedaan signifikan antara tipe CNGA3 dan CNGB3, sedangkan tipe GNAT2 relatif mempertahankan kerucut2).
Tes penglihatan warna: Pada tabel Ishihara, hampir semua tabel tidak terbaca kecuali tabel demonstrasi. Pada Panel D-15, menunjukkan pola kesalahan pada sumbu skotopik (antara deutan dan tritan). Pada anomaloskop, menunjukkan kemiringan curam dan tidak mencakup rentang kesetaraan warna normal.
QApakah ketajaman penglihatan pada buta warna total memburuk seiring bertambahnya usia?
A
Ketajaman visual terbaik terkoreksi seringkali stabil dalam jangka panjang. Di sisi lain, evaluasi struktural dengan OCT dapat menunjukkan perubahan seiring waktu (kerusakan zona elipsoid, perluasan ruang optik kosong) yang dapat berkembang3). Terjadi disosiasi antara fungsi dan struktur, dan pemantauan rutin melalui pemeriksaan berkala sangat penting.
Pewarisan autosomal resesif. Jika kedua orang tua adalah karier (pembawa), risiko anak terkena penyakit adalah 25%1). Sering terjadi pada anak dari orang tua yang tampak sehat, dan dalam banyak kasus tidak ada riwayat keluarga.
Pola pewarisan non-Mendelian akibat uniparental disomi (UPD) dari pihak ayah juga telah dilaporkan. Pada kasus homozigot untuk CNGA3 c.778G>C (p.D260H) yang terbentuk melalui UPD, tidak ditemukan mutasi pada ibu 6). Contoh-contoh seperti ini memiliki arti penting dalam penilaian risiko kekambuhan pada konseling genetik.
Pola mutasi: Mutasi missense dominan. Domain transmembran S4 merupakan titik panas
Distribusi geografis: Di Timur Tengah dan Cina, CNGA3 mencapai lebih dari 80%
CNGB3
Kromosom: 8q21.3
Fungsi: Subunit beta saluran CNG
Frekuensi: Sekitar 50% kasus1)2)
Pola mutasi: Mutasi nonsense, frameshift, dan splicing dominan. c.1148delC adalah mutasi paling sering
Distribusi geografis: Di Eropa dan Amerika, CNGB3 mencapai lebih dari 50%
Gen Lainnya
GNAT2 (1p13.3): Transdusin α kerucut. Sekitar 2%. Relatif ringan dengan lapisan fotoreseptor yang terpelihara 2)
PDE6C (10q23.33): Subunit α PDE kerucut. Tipe berat dengan onset dini 5)
PDE6H (12p12.3): Subunit γ PDE kerucut. Sangat jarang 1)
ATF6 (1q23.3): Faktor transkripsi respons stres retikulum endoplasma. Sekitar 2%. Mekanisme yang tidak terlibat langsung dalam transduksi cahaya 1)2)
Alel penetrasi rendah: CNGB3 c.1208G>A (p.R403Q) mempertahankan fungsi parsial, sehingga menghasilkan fenotip ringan 2).
Karakteristik mutasi PDE6C: Pada 4 kasus dengan mutasi baru (c.1670G>A, c.2192G>A), triad (nistagmus, fotofobia, gangguan penglihatan warna) ditemukan pada semua kasus. Elektroretinogram menunjukkan hilangnya penglihatan siang dan flicker 30 Hz dengan penglihatan malam normal, dan heterozigot majemuk menunjukkan fenotip yang lebih berat 5).
Warisan digenik: Terdapat kasus langka dengan mutasi pada kedua CNGA3 dan CNGB3 1).
QMengapa anak bisa menderita buta warna total meskipun kedua orang tua tidak menderita?
A
Karena buta warna total adalah penyakit resesif autosomal, kedua orang tua mungkin masing-masing membawa satu salinan gen mutan (pembawa) tanpa gejala dan dengan penglihatan warna normal. Jika dua pembawa memiliki anak, probabilitas anak mewarisi dua salinan mutasi dan menderita penyakit adalah 25% 1).
Jika nistagmus, fotofobia, dan penurunan penglihatan diamati dalam beberapa minggu pertama setelah lahir, diperlukan pemeriksaan komprehensif dengan mempertimbangkan kemungkinan ACHM. Pemeriksaan genetik sangat penting untuk diagnosis pasti.
Elektroretinogram
Signifikansi diagnostik: Standar emas
Temuan tipe komplit: Respons kerucut pada elektroretinografi fotopik menghilang atau sangat berkurang. Respons batang pada elektroretinografi skotopik normal hingga hampir normal1)2)
Temuan tipe inkomplit: Respons kerucut lemah yang sesuai dengan fungsi kerucut residual terdeteksi
Karakteristik tipe GNAT2: Kerucut S relatif lebih terawetkan dibandingkan tipe CNGA3/CNGB3
OCT
Signifikansi diagnostik: Standar untuk evaluasi struktural
Konten evaluasi: Stadium 5 tingkat lapisan luar fovea, deteksi hipoplasia fovea2)3)
Follow-up: Meskipun stabil secara fungsional, progresi struktural dapat terjadi, sehingga pemantauan rutin penting
Pemeriksaan tambahan: Evaluasi multimodal yang menggabungkan FAF dan AOSLO
Tes Genetik
Signifikansi diagnostik: Penting untuk diagnosis pasti
Metode: Panel target untuk 6 gen (CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)2)
Tingkat resolusi: Gen penyebab dapat diidentifikasi pada lebih dari 90% kasus1)
Partisipasi uji klinis: Diagnosis molekuler wajib untuk berpartisipasi dalam uji klinis terapi gen
Meskipun hiperopia sering terjadi, terdapat berbagai kelainan refraksi, sehingga koreksi dengan kacamata atau lensa kontak penting untuk memaksimalkan ketajaman penglihatan. Jika terdapat ambliopia, pertimbangkan terapi oklusi atau atropin.
Lensa pelindung cahaya untuk mengurangi fotofobia secara signifikan meningkatkan kualitas hidup sehari-hari. Data survei pasien menunjukkan 96% lebih memilih filter abu-abu daripada merah, dan 74% lebih memilih filter abu-abu di luar ruangan2). Pemilihan yang disesuaikan dengan preferensi individu dan lingkungan aktivitas pasien sangat penting.
Diperlukan penjelasan dan dukungan dari ahli mengenai karakteristik pewarisan resesif autosomal, risiko kekambuhan, dan pentingnya diagnosis genetik. Disarankan juga untuk menjalani diagnosis genetik sebagai persiapan untuk berpartisipasi dalam uji klinis terapi gen di masa depan.
QWarna apa yang cocok untuk kacamata pelindung cahaya?
A
Dalam survei pasien, 96% lebih memilih filter abu-abu daripada filter merah, dan 74% lebih memilih filter abu-abu di luar ruangan 2). Namun, karena perbedaan individu, disarankan untuk mencoba berbagai filter dan memilih yang sesuai dengan lingkungan aktivitas Anda dengan berkonsultasi dengan dokter mata atau ahli low vision.
6. Patofisiologi dan Mekanisme Penyakit yang Mendetail
Kaskade transduksi sinyal cahaya pada sel kerucut normal adalah sebagai berikut1).
Dalam gelap: Konsentrasi cGMP intraseluler tinggi, saluran CNG terbuka, Na⁺ dan Ca²⁺ masuk, sel mempertahankan keadaan depolarisasi, dan melepaskan glutamat secara terus-menerus.
Mutasi CNGA3: Mutasi missense mengganggu pelipatan protein, transportasi intraseluler, dan integrasi membran1)7). Domain transmembran S4 merupakan titik panas mutasi. Lebih dari 150 mutasi missense telah dilaporkan, 103 mutasi belum ditentukan patogenisitasnya, tetapi analisis struktur-fungsi 3D menunjukkan bahwa 86,4% memiliki konsekuensi fungsional yang mirip dengan mutasi patogen yang diketahui7).
Mutasi CNGB3: Mutasi nonsense dan pergeseran kerangka menghasilkan protein saluran terpotong atau kehilangan fungsi1). Saat CNGB3 defisien, saluran homomer CNGA3 masih tersisa, sehingga mungkin mempertahankan fungsi kerucut yang minimal.
Mutasi ATF6: Merupakan faktor transkripsi yang terlibat dalam respons protein tidak terlipat (UPR) retikulum endoplasma, dan tidak terlibat langsung dalam kaskade transduksi cahaya1)2). Mekanisme patologisnya berbeda dari gen lain, dan masih terus diteliti.
Saluran CNG memiliki struktur tetramer (CNGA3 × 3 dan CNGB3 × 1, pada beberapa laporan 2:2), setiap subunit memiliki 6 domain transmembran, domain pengikat nukleotida siklik, daerah penghubung C, dan domain pembentuk pori1).
Perkembangan utama kerucut pascakelahiran dan morfologinya mendekati normal, dan degenerasi diyakini dimulai pada awal masa dewasa. Akumulasi cGMP terlibat dalam proses degenerasi, dan perkembangan yang lebih cepat di daerah kaya kerucut S telah ditunjukkan pada model hewan1).
Secara tradisional, ACHM dianggap sebagai penyakit non-progresif. Namun, observasi jangka panjang dengan OCT menunjukkan bahwa meskipun ketajaman visual terbaik terkoreksi relatif stabil, perubahan struktural (penghancuran EZ, perluasan celah optik) terus berkembang3). Disosiasi fungsi-struktur ini penting dalam mengevaluasi jendela terapi untuk terapi gen.
Di zona bebas batang (area padat kerucut) di fovea, kehilangan kerucut sangat menonjol, sementara di parafovea, batang mengkompensasi fungsi dengan berkontribusi pada pita EZ3).
7. Penelitian Terbaru dan Prospek Masa Depan (Laporan Tahap Penelitian)
Pada uji coba NCT03001310 (AAV8-hCARp.hCNGB3) yang melibatkan 11 dewasa dan 12 anak-anak (total 23 orang), keamanan dapat diterima. Perbaikan penglihatan warna terjadi pada 6/23, perbaikan fotofobia pada 11/20, dan perbaikan kualitas hidup pada 21/23. Pada dosis tinggi, teramati kecenderungan peningkatan peradangan intraokular 2).
Pada uji coba AAV8.CNGA3 (RD-CURE), 9 orang menerima tiga dosis (1×10¹⁰ hingga 1×10¹¹ vg/mata). Data 1 tahun dan 3 tahun menunjukkan kecenderungan perbaikan ketajaman penglihatan dan sensitivitas kontras, tetapi tidak mencapai signifikansi statistik. Keamanan baik 1)2).
McKyton dkk. (2021) melakukan pemetaan kortikal dengan fMRI setelah injeksi subretinal AAV2tYF-PR1.7-hCNGA3 (NCT02935517) pada dua orang dewasa dengan CNGA3-ACHM 4). Mata yang diobati menunjukkan toleransi cahaya 5 kali lebih besar dibandingkan mata yang tidak diobati (perbaikan dramatis fotofobia), memperoleh kemampuan deteksi merah, dan pengecilan ukuran population receptive field (pRF) yang menunjukkan peningkatan resolusi spasial. Di sisi lain, tidak terlihat aktivasi area kortikal spesifik warna (misalnya V4), dan elektroretinogram lapangan penuh masih tidak mendeteksi respons kerucut. Pasien melaporkan perbaikan kehidupan sehari-hari seperti “peningkatan rasa aman saat menyeberang jalan”, “tidak perlu kaca pembesar”, dan “tidak perlu kacamata hitam di luar ruangan”.
Pemulihan fungsi setelah penggantian gen telah dikonfirmasi pada beberapa model hewan 1)2).
Model tikus: Pemulihan hingga 80% dari normal pada elektroretinogram.
Model anjing (CNGB3): Pemulihan elektroretinogram kerucut flicker setelah tindak lanjut 2,5 tahun pasca penggantian gen. Perbaikan perilaku pada lingkungan >25 lux.
Model domba (CNGA3): Perbaikan jangka panjang setidaknya 6 tahun setelah penggantian gen.
Mengenai jendela waktu efektif terapi, percobaan pada hewan menunjukkan bahwa pengobatan pada usia muda lebih efektif. Tikus tua menunjukkan respons yang buruk, namun pretreatment dengan CNTF (faktor neurotropik siliaris) memungkinkan pemulihan fungsi pada anjing tua 1)2).
Uji coba klinis (NCT04041232) untuk fenilbutirat gliserol (PBA) yang menargetkan respons stres retikulum endoplasma sedang berlangsung 2). Pendekatan ini menarik perhatian sebagai alternatif penggantian gen dengan mengurangi misfolding di retikulum endoplasma.
Tantangan utama terapi gen adalah sebagai berikut 1)2)4).
Imunogenisitas: Respons imun terhadap kapsid AAV dapat membatasi efektivitas terapi.
Optimalisasi dosis: Diperlukan penyeimbangan antara efektivitas terapi dan risiko peradangan intraokular.
Tantangan perkembangan: Kesulitan operasi pada mata anak-anak, penanganan munculnya ambliopia.
Keterbatasan plastisitas korteks visual: Pada orang dewasa, reorganisasi fungsional korteks mungkin sulit, sehingga waktu terapi menjadi penting.
Efektivitas jangka panjang dan biaya: Menjamin efek yang berkelanjutan dan tantangan ekonomi kesehatan.
QDapatkah terapi gen menyembuhkan buta warna total secara sempurna?
A
Saat ini, masih dalam tahap uji keamanan dan kemanjuran fase I/II. Pengurangan fotofobia dan perbaikan sebagian sensitivitas cahaya telah dilaporkan, namun pemulihan penglihatan warna secara penuh belum tercapai2)4). Penelitian pada hewan menunjukkan bahwa pengobatan pada usia muda mungkin efektif, tetapi data jangka panjang pada manusia masih terbatas. Penting untuk memantau kemajuan penelitian dan memperbarui informasi secara teratur dengan dokter yang merawat.
McKyton A, Averbukh E, Marks Ohana D, Levin N, Banin E. Cortical Visual Mapping following Ocular Gene Augmentation Therapy for Achromatopsia. J Neurosci. 2021;41(35):7363-7371. doi:10.1523/JNEUROSCI.3222-20.2021. PMID:34349002; PMCID:PMC8412991.
Madeira C, Godinho G, Grangeia A, Falcão M, Silva R, Carneiro Â, et al. Two Novel Disease-Causing Variants in the PDE6C Gene Underlying Achromatopsia. Case reports in ophthalmology. 2021;12(3):749-760. doi:10.1159/000512284. PMID:34720973; PMCID:PMC8460892.
Kohl S, Baumann B, Dassie F, et al. Paternal Uniparental Isodisomy of Chromosome 2 in a Patient with CNGA3-Associated Autosomal Recessive Achromatopsia. Int J Mol Sci. 2021;22(15):7842. doi:10.3390/ijms22157842. PMID:34360608; PMCID:PMC8346044.
Rasmussen DK, Sun YJ, Franco JA, et al. Structure-function analysis of CNGA3-associated achromatopsia patient variants complements clinical genomics in pathogenicity determination. Orphanet J Rare Dis. 2025;20:261. doi:10.1186/s13023-025-03792-3.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.